Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(2):181-190. doi:10.7150/ijms.17119 This issue Cite
Research Paper
Immune Cell Repertoire and Their Mediators in Patients with Acute Myocardial Infarction or Stable Angina Pectoris
Wenwen Yan1, Yanli Song2, Lin Zhou1, Jinfa Jiang1, Fang Yang3, Qianglin Duan1, Lin Che1, Yuqin Shen1 ![]() , Haoming Song1
, Haoming Song1 ![]() , Lemin Wang1
, Lemin Wang1 ![]()
1. Department of Cardiology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China;
2. Department of Emergency Medicine, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China;
3. Department of Experimental Diagnosis, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China.
Received 2016-8-5; Accepted 2016-12-21; Published 2017-2-8
Abstract
Background: To evaluate the natural innate and adaptive immunity through gene expression and cytology levels in peripheral blood mononuclear cells in patients with acute myocardial infarction (AMI), stable angina pectoris (SAP) and controls.
Methods: 210 patients with AMI, 210 with SAP, and 250 clinical controls were recruited. Whole human genome microarray analysis was performed in 20 randomly chosen subjects per group were examined to detect the expressions of complement markers, natural killer cells, T cells and B cells. The quantity of these cells and related cytokines as well as immunoglobulin levels were measured in all subjects.
Results: In AMI group, the mRNA expressions of late complement component, markers of natural killer cells, CD3+, CD8+ T cells and B cells were down-regulated, while those of early complement component and CD4+T cells were up-regulated (p<0.05). In both AMI and SAP patients, the quantity of natural killer cells, CD3+, CD8+ T cells, B cells, IgM and IgG were significantly lower than those of the controls. CD4+ T cells, CH50, C3, C4, IL-2, IL-4, IL-6 and IFN-γ were significantly higher (p<0.05).
Conclusions: In AMI patients, both of gene expressions related to complement, natural killer cells, CD3+, CD8+ T cells, B cells and the quantity of these immune cells decreased while cell number reduced in SAP patients. Immune function in both AMI and SAP patients decreased especially in AMI patients with declined gene and protein levels. To improve the immune system is a potential target for medical interventions and prevention in AMI.
Keywords: myocardial infarction, stable angina pectoris, gene expression, innate immunity, adaptive immunity.
Introduction
Cardiovascular diseases (CADs), with high morbidity and mortality worldwide, are caused mainly by atherosclerosis. In particular, acute myocardial infarction (AMI) represents life-threatening conditions during the history of CAD [1, 2]. Nowadays we are still unable to effectively predict and prevent AMI occurrence. The pathologic mechanism responsible for majority of AMI is the rupture of stable atherosclerotic plaque and thrombosis [3]. Obviously, there must be a trigger to induce the sudden rupture. Infection seems to be undoubtedly linked to vulnerable atherosclerotic lesion; however, its role cannot be easily documented [4, 5, 6]. Various exogenous microorganism infections, including Chlamydia pneumoniae, Helicobacter pylori, Cytomegalovirus and Bacteroides gingivalis are accepted as the new susceptible factors of CAD [7, 8].
Our recent study demonstrated the decreased T cell immunity function in AMI patients [9, 10]. T cells, as a key component of adaptive immune system, eliminate the pathogenic microorganisms and malignant cells. The significant decline of T cell function suggests that the pathogenesis of acute thrombosis in AMI patients may be associated with the depletion of immune cells. However, less is known about the nature of immune response in different stages of CAD [11, 12]. In recent study, we designed this in vitro study to investigate both innate and adaptive immunity in patients with AMI or stable angina pectoris (SAP). Human microarray analysis was used to systematically measure them RNA expression of the complement component, markers of immune cells in peripheral blood mononuclear cells (PBMCs) from AMI, SAP and controls. Moreover, the quantity of immune cells, related cytokines and immunoglobulin levels were also measured.
Material and Methods
Patients' Information
The study recruited 210 patients with AMI, 210 with SAP, and 250 clinically controls. Human microarray analysis was performed in 20 randomly chosen subjects per group. The sample sizes and the number of subjects per group were based on an assumed within-group variance of 0.50 and the targeted nominal power of 0.95 [13]. Table 1 shows the baseline demographic data. All patients were enrolled between Mar 2013 and Feb 2015 from our Coronary Care Unit and Cardiovascular Department. The AMI patients were admitted no more than 12 hours from the onset of symptoms to our Coronary Care Unit including 180 males and 30 females, with an average age of 59±11 years. The SAP group included 210 patients (176 males, 34 females, aged 64±11 years). 250 healthy volunteers (207 males, 43 females, aged 61± 9 years) were enrolled as the control group during the same period. Histories, physical examination, ECG, chest radiography and routine chemical analyses showed the controls had no evidence of coronary heart diseases.
Baseline demographic data in three groups (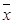 ± s.d.).
± s.d.).
| Index | AMI (a) (N=210) | SAP (b) (N=210) | Con(c) (N=250) | P (all) | P (a v b) |
|---|---|---|---|---|---|
| Age | 58.5±10.7 | 63.6±11.1 | 60.9 ± 9.4 | 0.141 | 0.211 |
| Sex(M/F) | 180/30 | 176/34 | 207/43 | 0.694 | 0.773 |
| BMI(kg/m2) | 24.6±2.9 | 22.5±2.2 | 22.7±1.9 | 0.112 | 0.76 |
| Ethnicity, Han | 210 | 210 | 250 | 1 | 1 |
| Tobacco smoking(num/d) | 13.6±10.1 | 14.4±8.4 | 11.2±6.1 | 0.24 | 0.648 |
| SBP (mmHg) | 130.1±11.3 | 123.7±10.1 | 124.8±7.8 | 0.145 | 0.701 |
| DBP (mmHg) | 67.7±8.8 | 72.0±8.8 | 77.6±3.6 | 0.126 | 0.24 |
| LDL-C(mmol/L) | 2.8±1.2 | 2.4±1.8 | 2.7±1.5 | 0.44 | 0.676 |
| Triglycerides(mmol/L) | 1.5±1.8 | 1.7±1.0 | 1.8±0.7 | 0.51 | 0.12 |
| HDL-C(mmol/L) | 0.7±0.9 | 0.8±0.7 | 0.9±0.2 | 0.11 | 0.303 |
| FBG (mmol/L) | 5.3±0.4 | 5.1±0.7 | 5.0±0.2 | 0.24 | 0.834 |
Footnotes: BMI= body mass index; SBP=systolic blood pressure; DBP =diastolic blood pressure; LDL-C=low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol; FBG: Fasting Plasma Glucose.
All AMI patients were diagnosed on the basis of following criteria [14]: Detection of a rise of cardiac biomarker values [preferably cardiac troponin (cTn)] with at least one value above the 99th percentile upper reference limit (URL) and with at least one of the following: 1) Symptoms of ischemia. 2) New or presumed new significant ST-segment-T wave (ST-T) changes or new left bundle branch block (LBBB). 3) Development of pathological Q waves in ECG. 4) Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.5) Identification of an intracoronary thrombus by angiography.
All SAP patients had exclusively effort-related angina with a positive exercise stress test and at least one coronary stenosis was detected at angiography (>70% reduction of lumen diameter).
There were no significant differences among three groups in age, sex, body mass index (BMI), ethnicity, smoking status, systolic blood pressure (SBP), diastolic blood pressure (DBP), low-density lipoprotein cholesterol (LDL-C), triglycerides, high-density lipoprotein cholesterol (HDL-C) and fasting plasma glucose (FBG) (Table 1).
The exclusion criteria for three groups were as follows: venous thrombosis, history of severe renal or hepatic diseases, hematological disorders, acute or chronic inflammatory diseases and malignancy.
The study protocol was approved by the ethics committee of Tongji University and informed consent form was obtained.
Gene Expression Chips
Agilent G4112F Whole Human Genome Oligo Microarrays purchased from Agilent (USA) were used in the chip analysis. A microarray is composed of more than 41,000 genes or transcripts, including targeted 19,596 entrez gene RNAs. Sequence information used in the microarrays was derived from the latest databases of RefSeq, Goldenpath, Ensembl and Unigene [15]. More than 70% of the gene functions in the microarray are already known. All 20 randomly selected patients for each group were subjected to the chip analysis.
Total RNA Isolation
Ten milliliter of peripheral blood samples from median cubital vein were drawn from all the patients immediately after admission. Four milliliter blood was kept in PAXgene tube for total RNA isolation and the rest six milliliter was for laboratory assays. Leucocytes were obtained through density gradient centrifugation with Ficoll solution and the remaining red blood cells were destroyed by erythrocyte lysis buffer (Qiagen, Hilden, Germany). Following the manufacturer's instructions, total RNA was extracted and purified using PAXgeneTM Blood RNA kit (Cat#762174, QIAGEN, GmBH, Germany). We further checked for a RIN number to inspect RNA integration by an Agilent Bio analyzer 2100 (Agilent technologies, Santa Clara, CA, US). The sample was considered qualified when both 2100 RIN and 28S/18S were larger than or equal to 0.7.
RNA Amplification and Labeling
Total RNA was amplified and labeled by Low Input Quick Amp Labeling Kit, One-Color (Cat#5190-2305, Agilent technologies, Santa Clara, CA, US), following the manufacturer's instructions. Labeled cRNA was purified by RNeasy mini kit (Cat#74106, QIAGEN, GmBH, Germany).
Microarray Hybridization
Each slide was hybridized with 1.65ìg Cy3-labeled cRNA using Gene Expression Hybridization Kit (Cat#5188-5242, Agilent technologies, Santa Clara, CA, US) in Hybridization Oven (Cat#G2545A, Agilent technologies, Santa Clara, CA, US), following the manufacturer's instructions. After 17 hours of hybridization, slides were washed in staining dishes (Cat#121, Thermo Shandon, Waltham, MA, US) with Gene Expression Wash Buffer Kit (Cat#5188-5327, Agilent technologies, Santa Clara, CA, US), according to the manufacturer's operation manual.
Chip Scan and Data Acquisition
Slides were scanned using Agilent Microarray Scanner (Cat#G2565CA, Agilent technologies, Santa Clara, CA, US) with default settings. Dye channel: Green, Scan resolution=3ìm, 20bit. Data were extracted with Feature Extraction software 10.7 (Agilent technologies, Santa Clara, CA, US). Raw data were normalized using Quantile algorithm, Gene Spring Software 11.0 (Agilent technologies, Santa Clara, CA, US).
RT-PCR
The spots in the microarray were randomly selected and their expressions were confirmed by RT-PCR. Among all the genes with different expressions, three genes were randomly selected and subjected to RT-PCR, along with the house keeping genes (GAPDH). The relative expressions were indicated as the expression of the target genes normalized to the expression of GAPDH (2-ÄÄCt). The melting curve and the 2-ÄÄCt-method were used to detect the differences in the expressions among the three groups. The results from RT-PCR were consistent with the microarray analysis.
Laboratory assays
Two milliliter blood sample was anticoagulated with EDTA-K3 for the counting of CD16+CD56+ natural killer cells, T lymphocyte subsets and CD19+B cells, and the rest four milliliter was separated by centrifugation within 1 hour for the examination of serum immunoglobulin and cytokines. All tests were finished within two weeks.
CH50 was detected with liposome immune assay (Beckmann DxC-800 fully automatic biochemical analyzer, USA; Reagents: Wako Pure Chemical Industries, Ltd., Japan). C3 and C4 were detected with immunone-phelometry (BNII system, Siemens AG, Germany; Reagents: Siemens Healthcare Diagnostics Products GmbH, Germany).
Cytokines, including IL-2, IL-4, IL-6 and IFN-γ were measured by double antibody sandwich ELISA assay (Microplate reader Model 2010, Anthos, Austria; Reagents: Dili biotech, Shanghai). Serum levels of IgA, IgM and IgG were calculated by the immunonephelometric technique using the automated IMMAGE 800 immunochemistry system (Beckman Coulter, Brea, CA, USA), and expressed as g/L.
Leukocyte subpopulations were measured by flow cytometry (BEPICS XL-4, BECKMAN- COULTER). Monoclonal antibodies against CD3, CD4, CD8, CD16, CD56 and CD19 were purchased from BD Biosciences. The antibodies were marked with one of three fluorochromes: fluorescein isothiocyanate (FITC), phycoerythrin (PE) and phycoerythrin-cyanin 5.1 (PC5). Cells were identified by combinations as follows: CD3 (FITC)/CD16 (PE)/CD56 (PC5) (NK cells), CD3 (FITC)/CD4 (PE)/CD8 (PC5) (CD4andCD8 cells), and CD19 (PE) (B cells). In brief, 100 μL of EDTA treated blood was added to each tube and control tube was also included. 20 μL of mouse IgG1-FITC, IgG1-PE or IgG1-PC5 was then added, followed by addition of corresponding fluorescence antibodies. Following vortexing, incubation was done in dark for 30 min at room temperature. 500 μL of hemolysin (BECKMAN-COULTER) was then added, followed by incubation at 37°C for 30 min. Following washing, 500 μL of sheath fluid was added to each tube, followed by flow cytometry (EPICS XL-4, BECKMAN-COULTER). The PMT voltage, fluorescence compensation and sensitivity of standard fluorescent microspheres (EPICS XL-4, BECKMAN-COULTER) were used to adjust the flow cytometer and a total of 10,000 cells were counted for each tube. The corresponding cell population in the scatter plot of isotype controls was used to set the gate, and the proportion of positive cells was determined in each quadrant (%). SYSTEM-II was used to process the data obtained after flow cytometry.
Statistical Analysis
Descriptive statistics were expressed as mean ± s.d. Differences between groups were examined by one-way analysis of variance (ANOVA). After ANOVA the test of all pairwise group mean comparison was performed using the Tukey's method. Density curves for CH50, C3, C4, CD16+CD56+, CD3+, CD4+, CD8+and CD19+ cells were delineated using R software. Data were analyzed using SPSS 17.0, and p-values <0.05 were considered statistically significant.
Results
Gene expression and serum level of the complement
The results showed mRNA expressions of early and late complement components including C1qá, C1qâ, C1qɤ, C1r, C1s, C2, C3, C4b, C5a, C6, C7, C8á, C8â, and C8ɤ and C9 were examined in PBMCs from three groups of patients (Figure 1A). In AMI group, gene expressions of C1qá (p<0.05), C1qâ, C1qɤ, C1r and C5a were significantly up-regulated (all p<0.01), whereas expressions of C7, C8â and C9 were significantly down-regulated when compared with SAP patients and controls, respectively (p<0.05). C1s expression in AMI patients was significantly lower than the controls (p<0.05). Serum CH50, C3 and C4 levels were significantly increased in AMI and SAP patients when compared with controls (p<0.01). CH50 was significantly higher in AMI patients than in SAP patients (p<0.01). There was no significant difference between AMI and SAP patients in C3 and C4 levels. The density curves of CH50, C3 and C4 are shown in Figure 1B-D separately.
From three groups in PBMCs, (A) mRNA expression of early and late components complement. (B) Serum CH50 level. (C) Serum C3 level. (D) Serum C4 level. Three groups: *, P<0.05; **, P<0.01. AMI vs. Con: #, P<0.05; ##, P<0.01. AMI vs. SAP: +, P<0.05; ++, P<0.01. SAP vs. Con &: P<0.05; &&: P<0.01.
Gene expression and counting of NK cells
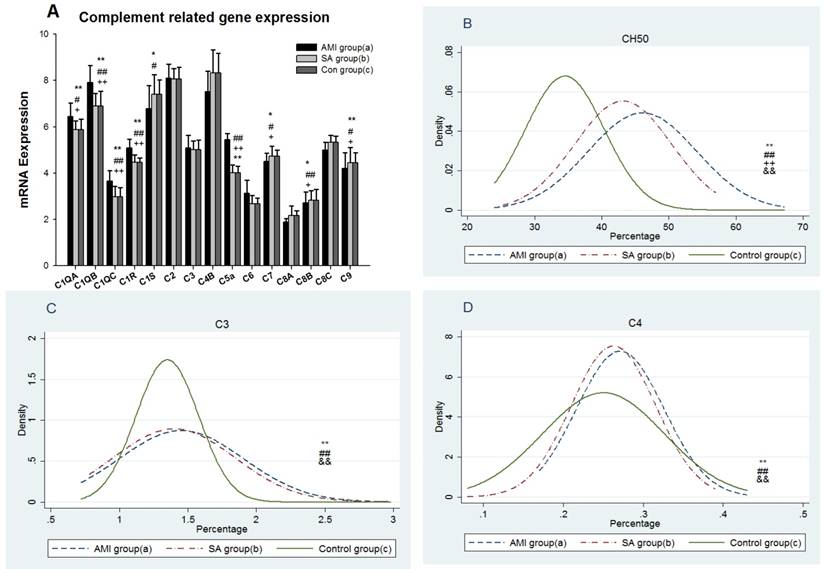
The results showed 12 gene expressions of NK cell biomarkers[16], including CD16, CD56, five inhibitory receptors,CD94, NKG2A, CD158(KIR2DL), CD161(KLRB1), CD328( Siglect-7) and five activating NK cell receptors, including CD335(Nkp46), CD337(Nkp30), CD48(2B4), CD314(NKG2D) and CD319(CRACC) in PBMCs from three groups (Figure 2A). In AMI group mRNA expressions of the genes encoding CD94, NKG2A, CD158, CD161, CD337, CD314 and CD319 were significantly lower than in SAP patients and the controls (p<0.05). There was no statistical difference in NK cell biomarker expressions between SAP and the controls. Density curves for the NK cell proportion in PBMCs from three groups were delineated (Figure 2B). The two density curves of cell proportion from AMI and SAP patients in PBMCs were substantially left shift when compared with the controls. The number of NK cells was significantly decreased in both AMI and SAP patients (p<0.01). However, there was no significant difference between AMI and SAP patients in the quantity of NK cells.
From three groups in PBMCs, (A) mRNA expression of intracellular and extracellular markers of CD16+CD56+cells. (B) The comparison of CD16+CD56+ cells counting. Three groups: *, P<0.05; **, P<0.01. AMI vs. Con: #, P<0.05; ##, P<0.01. AMI vs. SAP: +, P<0.05; ++, P<0.01. SAP vs. Con &: P<0.05; &&: P<0.01.
Gene expression, subsets counting and related cytokines of T cells
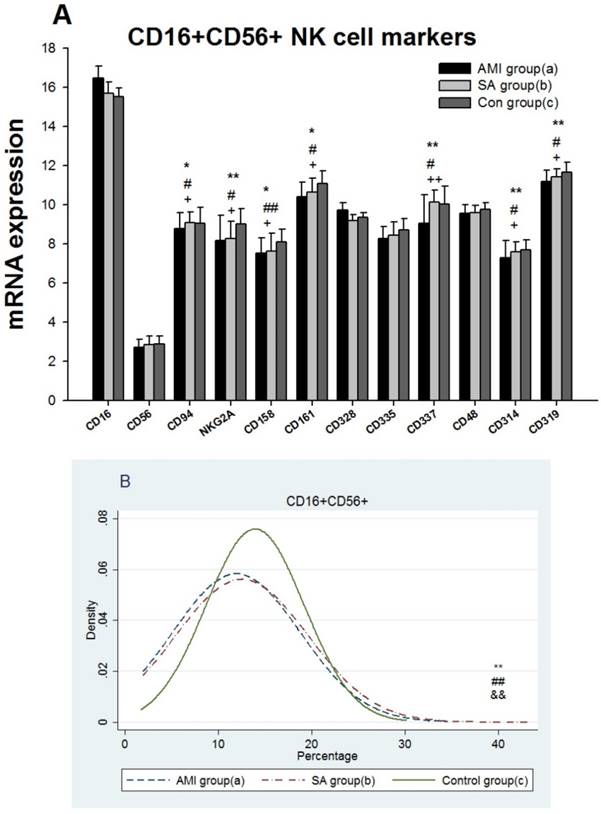
Expressions of 8 genes related to T cell receptor (TCR) antigen recognition, 16 genes associated with CD4+T cells and 15 genes with CD8+ T cells were detected among three groups (Figure 3A, 3C, 3E). 16 genes in AMI patients encoding TCRA, TCRB, TCRG, TCRZ, CD3D, CD3E, CD3G, CD195(CCR5), IL-10, GATA3, CD278(ICOS), CD8A, CD8B, CD28, GZMM and CASP10 were significantly down-regulated when compared with the SAP patients and controls respectively(p<0.05). TCRIM, CD294 (CRTH2) and GZMK expressions in AMI group were significantly lower than those in SAP group (p<0.05). Comparing with controls, gene expressions of CD4, IL4 and TNFA in AMI group were significantly down-regulated (p<0.05), while IL-2 and CD366 (Tim-3) mRNA expressions were up-regulated (p<0.05). Between SAP and control group, there was no statistical difference in TCR, CD4+ and CD8+T cell markers related mRNA expression.
From three groups in PBMCs, (A) Expression of genes related to T cell antigen recognition. (B) CD3+ counting. (C) Expression of genes related to CD4+. (D) CD4+ counting. (E) Genes related to CD8+. (F) CD8+ counting. Three groups: *, P<0.05; **, P<0.01. AMI vs. Con: #, P<0.05; ##, P<0.01. AMI vs. SAP: +, P<0.05; ++, P<0.01. SAP vs. Con &: P<0.05; &&: P<0.01.
Results from the proportions of cytological T lymphocyte subsets in PBMCs among three groups showed the levels of CD3+ and CD8+T cells in AMI and SAP group decreased significantly (p< 0.05), while CD4+T cells increased (p<0.01) when compared with control group (Figure 3B, 3D, 3F, Table 2). The cytokine IL-2, IL-4, IL-6 and IFN were significantly increased in AMI and SAP patients when compared with the controls (p<0.01). However, there was no significant difference between AMI and SAP patients in IL-2, IFN, CD3+ T cell and CD8+T cell quantity (Table 2). The counting of CD4+ T cell, IL-4 and IL-6 were higher in AMI patients than in SAP patients (p<0.01).
Values of T cell immunity among three groups (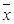 ± s.d.).
± s.d.).
| Index | AMI (a) (N=210) | SAP (b) (N=210) | Con(c) (N=250) | P (all) | P (a/c) | P (b/c) | P (a/b) |
|---|---|---|---|---|---|---|---|
| CD3+ (%) | 66.7±10.7 | 68.4±10.0 | 71.5±9.2 | 0.00 | 0.00 | 0.002 | 0.275 |
| CD4+ (%) | 42.1±8.9 | 45.1.±9.2 | 37.0±9.1 | 0.00 | 0.00 | 0.00 | 0.003 |
| CD8+ (%) | 21.8±7.1 | 23.3±6.7 | 28.6±6.9 | 0.00 | 0.00 | 0.00 | 0.068 |
| IL-2 (pg/ml) | 34.2±18.5 | 33.5±14.5 | 9.9±2.3 | 0.00 | 0.00 | 0.00 | 0.96 |
| IL-4(pg/ml) | 35.7±15.7 | 28.22±10.9 | 4.8±2.3 | 0.00 | 0.00 | 0.00 | 0.00 |
| IL-6 (pg/ml) | 29.9±16.2 | 24.6±14.4 | 3.0±1.4 | 0.00 | 0.00 | 0.00 | 0.001 |
| IFN (pg/ml) | 40.8±21.4 | 33.0±22.1 | 16.7±6.3 | 0.00 | 0.00 | 0.00 | 0.72 |
Gene expression, counting and serum immunoglobulin level of B cells
The results showed that expressions of 15 genes related to B cell biomarkers in patients with AMI, SAP and the controls (Figure 4A), including CD5, CD19, CD20, CD21 (CR1), CD22, CD23, CD40, CD79a, CD79b, CD80(B7-1), CD86(B7-2), CD138, CD154(IgM), CD268(BAFFR) and CD279(PD-1). In PBMCs from three groups, expressions of 8 genes encoding CD5, CD19, CD20, CD22, CD40, CD79b, CD268 and CD279 in AMI group were significantly lower than those from SAP and control group (p<0.05). Compared with controls, gene expressions of CD21, CD23 and CD79a were significantly down-regulated in AMI patients (p<0.05). Between the SAP and control group, there were no significant differences in B cell marker expressions. When compared with controls, B cell counting, IgG and IgM in PBMCs were significantly down-regulated (p<0.01), while IgA was significantly increased in both AMI and SAP group (Figure 4B, Table 3) (p<0.05).
From three groups in PBMCs, (A) mRNA expression of intracellular and extracellular markers of CD19+ cell. (B) The comparison of CD19+ cells counting. Three groups: *, P<0.05; **, P<0.01. AMI vs. Con: #, P<0.05; ##, P<0.01. AMI vs. SAP: +, P<0.05; ++, P<0.01. SAP vs. Con &: P<0.05; &&: P<0.01.
Values of B cell immunity among three groups ( ± s.d.).
± s.d.).
| Index | AMI(a) (N=210) | SAP (b) (N=210) | Con(c) (N=250) | P (all) | P (a/c) | P (b/c) | P (a/b) |
|---|---|---|---|---|---|---|---|
| CD19+(%) | 9.4±5.0 | 10.3±4.7 | 13.7±4.6 | 0.00 | 0.00 | 0.00 | 0.212 |
| IgA (g/L) | 2.3±1.0 | 2.2±0.8 | 1.9±0.7 | 0.00 | 0.001 | 0.014 | 0.487 |
| IgM(g/L) | 0.8±0.42 | 0.8±0.35 | 1.2±0.41 | 0.00 | 0.00 | 0.00 | 0.572 |
| IgG (g/L) | 10.8±2.6 | 11.2±2.2 | 12.0±2.3 | 0.00 | 0.00 | 0.001 | 0.242 |
Discussion
In our current study, the significantly up-regulated mRNAs expressions of early complement components, C1qá, C1qâ, C1qɤ, C1r and C5a demonstrated that the classical pathways were activated in AMI patients. The initiation of classical pathway eventually results in the terminal access to form C5b-9 complex, which makes a transmembrane pore in the target cells' membrane to lysis [17]. C5b initiates the formation of MAC, which consists of C5b, C6, C7, C8, and multiple molecules of C9. In our study the significantly lowest levels of C7, C8â and C9 mRNAs in AMI patients suggested the obstacle of MAC formation. In AMI and SAP patients, the serum levels of C3, C4 and CH50, which reflected the activities of C1-C9 via classic pathway, were all elevated. Gene and cytology levels of the complement in both AMI and SAP patients were activated and the results were consistent with previous studies [18, 19, 20]. Though the complement was activated in AMI and SAP stages, based on the genomics results of complement cascade reaction imbalance, cytolytic effect of the complement only decreased in AMI patients.
NK cells express an array of inhibitory and activating receptors. The inhibitory receptors are responsible for self-tolerance while activating receptors mediate the NK cell cytotoxicity (NKCC) [21, 22]. KIRs are the most important NK cell receptors, including CD94, NKG2A, CD158 and CD314, which recognize classical MHC class I [23]. In present study, the gene expressions of CD94, NKG2A and CD158 were significantly lower than those in SAP patients and the controls, suggesting the impaired ability to protect normal cells in AMI patients. Receptors CD335 (NKp46), CD337 (NKp30), CD48 (2B4), CD314(NKG2D) and CD319 (CRACC) are most central activating receptors and play an important role in targeting NK cell responses toward abnormal cells and eventually the cell lysis [24, 25, 26, 27]. In our current study, gene expressions of activating receptors, CD337, CD314 and CD319 in AMI patients were significantly decreased in comparison with SAP patients and controls respectively, which showed the transduction of activating signal was inhibited in patients with AMI. The cytotoxic ability of NK cells was decreased afterwards. There was no significant difference in mRNA expression between the SAP patients and controls in inhibitory and activating receptors, indicating the NK receptors in SAP patients was in a nearly inactive state. Previous studies found the reduced proportions of NK cells in peripheral blood of CAD, but the reason was still controversial [28, 29, 30, 31]. The similar loss of NK cell numbers in both AMI and SAP patients were also observed in our study (Figure 2B). Together with the notably decreased expression of NK cell biomarkers in AMI patients, different levels of reduced immunity in NK cells were demonstrated in AMI and SAP stages. In AMI patients both numbers and receptor activity were decreased, while only a deficit of quantity was found in SAP patients.
TCR is a molecule found on the surface of T lymphocytes that is responsible for recognizing antigens. The first signal for T cell activation is provided through the TCR-CD3 [32]. In present study, gene expressions of TCRA, TCRB, TCRG, TCRZ, CD3D, CD3E and CD3G were significantly lower in AMI group than those in SAP and control group (Figure 4A), indicating the decreased ability of TCR antigen recognition. In addition, the loss of CD3+ T cells in PBMCs was found in both AMI and SAP patients (Figure 4B), suggesting the dysfunction of CD3+T cells in CAD, especially in AMI stage.
Naive CD4+ T cells differentiate into T helper type 1 (Th1) and T helper type 2 (Th2). Th1 cells achieve cellular immunity mainly by secreting IL2, IL12 and IFN-ã. T-bet is a Th1 transcription factor for regulating Th1 development [33]. CD195 (CCR5) and CD182 (CXCR3) are specific Th1 lymphocytes chemokine receptors [34]. Th2 cells produce IL4, IL6 and IL10 to activate B lymphocytes and generate antibodies. GATA3 is the Th2 specific transcription factor, and CCR3 together with CD294 (CRTH2) are chemokine receptors of Th2 cells [35, 36, 37]. CD366 (Tim-3) is a Th1-specific cell surface protein while CD365 (Tim-1) is Th2-specific [38, 39]. The high mRNA expressions of Th1 biomarkers (IL2 and CD366) and low RNA expressions of Th2 biomarkers (IL4, IL-10, CD278 and CD294) in AMI patients suggested a shift towards Th1 dominance. The significant increase of CD4+T cells, IL-4 and IL-6 in AMI than in SAP patients showed the differential degrees of CD4+ T cell mediated cellular immunity dysfunction in AMI and SAP patients.
CD8+ T cells kill virus-infected cells and tumor cells and play a critical role in immune protection [40]. CD8+T cell is firstly activated by TCR and CD8 binding and then co-stimulatory molecules.CD8+ T cells make the fatal attack through the perforin-granzyme, Fas-Fas ligand (FasL), and TNF-a pathways [41,42]. The presence of CD8+ T cells in atherosclerotic lesions is widely demonstrated but studies investigating their role in atherogenesis have yielded contradictory results [43, 44]. In the present study, all 15 genes related to killing ability of CD8+ T cells in AMI patients were down-regulated; especially CD8A, CD8B, CD28, CD278, GZMK, GZMM, PRF1 and CASP10 were significantly down-regulated when compared with SAP and/or controls. Together with significant loss of CD8+T cells in AMI and SAP patient in PBMCs indicated the decreased cytotoxic ability of CD8+ T cells in CAD patients, particularly in the stages of AMI patients.
We detected all 15 genes related with intracellular and extracellular markers of CD19+B cells [45] (Figure 4A). B cell receptor (BCR) was composed of membrane immunoglobulin (Ig) which recognizes the antigens while Igá (CD79A) and Igâ (CD79B) transmit the activation signals [46]. CD19 and CD21 are B cell co-receptors and enhance the BCR signal transduction [47]. The B cell specific Src-family kinase CD5, specifically binding B cell surface Ig, is dispensable for B cell activation [48]. CD268 is the principal receptor required for BAFF-mediated B cell activation [49]. CD279 encodes a cell surface membrane protein of Ig superfamily and plays a role in their differentiation [50]. In AMI patients, gene expressions of CD5, CD19, CD20, CD21, CD22, CD23, CD40, CD79A, CD79B, CD268 and CD279 were significantly lower than those in SAP and /or control group, which showed B cell activation were blocked in AMI patients. There was no significant gene expression difference in B cell activation between the SAP patients and the control group. The detection of B cell quantity, IgM and IgG levels in PBMCs were decreased in both AMI and SAP patients. In sum, in AMI patients, gene expressions and numbers of B cells were reduced, demonstrating the deeply weakened humoral immunity in AMI group.
In the present study, in AMI patients the mRNA expression of immune system was consistent with the cytological level and the decline of both parameters demonstrated the collapse of immune function in AMI group. In SAP patients, the immunity related gene expression was different from cytological level. The CH50, C3 and C4 were increased and the number of NK cells, CD3+, CD8+ T cells and CD19+ B cells were decreased, while the gene expression of immune system was in a nearly inactive status. In the current study, we can conclude that the attack of AMI and SAP was associated with different levels of immune dysfunction. AMI occurred in the stage of immune collapse while SAP occurred in progressively reduced level of immunity but still within the boundary of compensation. The quantity of immune cells in peripheral blood may reflect the current state of immune function and the gene expression of immune system stands for the compensatory capacity of the immune system.
In AMI patients, the suppressed innate and adaptive immune system, especially the cytotoxic ability, failed to remove the exogenous pathogens. Various exogenous microorganism infections are supposed as risk factors of AMI [8, 9] and infection seems to be linked to plaque rupture [4, 5, 6, 7].
Conclusions
The pathogenesis of AMI might be related with infections of pathogens under the depletion of immune system. That is the reason why single vaccine is ineffective on AMI prevention. To improve the immunity of CAD patients may be considered as a potential target for medical intervention and prevention of AMI.
Acknowledgements
The study was supported by Shanghai Traditional Chinese Medicine 3-year Development Program (2014-2016); National Natural Science Foundation (81570359) and Shanghai municipal health and Family Planning Commission project (20144Y0046).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317-325
2. Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481-3488
3. Shah PK. Pathophysiology of coronary thrombosis: role of plaque rupture and plaque erosion. Prog Cardiovasc Dis. 2002;44:357-368
4. Levi M, van der Poll T, Schultz M. New insights into pathways that determine the link between infection and thrombosis. Neth J Med. 2012;70:114-120
5. Chatzidimitriou D, Kirmizis D, Gavriilaki E. et al. Atherosclerosis and infection: is the jury still not in? Future Microbiol. 2012;7:1217-1230
6. Levi M, van der Poll T, Schultz M. Infection and inflammation as risk factors for thrombosis and atherosclerosis. Semin Thromb Hemost. 2012;38:506-514
7. Khan S, Rahman HN, Okamoto T. et al. Promotion of atherosclerosis by Helicobacter cinaedi infection that involves macrophage-driven proinflammatory responses. Sci Rep. 2014;4:4680
8. Mostafa A, Mohamed MK, Saeed M. et al. Hepatitis C infection and clearance: impact on atherosclerosis and cardiometabolic risk factors. Gut. 2010;59:1135-1140
9. Yan WW, Zhang KS, Duan QL. et al. Significantly reduced function of T cells in patients with acute arterial thrombosis. Journal of Geriatric Cardiology. 2015;12:287-293
10. Yan WW, Wang LM, Jiang JF. et al. Differential expression of T cell-related genes in AMI and SA stages of coronary artery disease. Int J ClinExp Med. 2015;8:10875-10884
11. Matusik P, Guzik B, Weber C. et al. Do we know enough about the immune pathogenesis of acute coronary syndromes to improve clinical practice? Thromb Haemost. 2012;108:443-456
12. Arbab-Zadeh A, Nakano M, Virmani R. et al. Acute coronary events. Circulation. 2012;125:1147-1156
13. Dobbin K, Simon R. Sample size determination in microarray experiments for class comparison and prognostic classification. Biostatistics. 2005;6:27-38
14. Thygesen K, Alpert JS, Jaffe AS. et al. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60:1581-1598
15. Wiltgen M, Tilz GP. DNA microarray analysis: principles and clinical impact. Hematology. 2007;12:271-287
16. Cooper MA, Colonna M, Yokoyama WM. Hidden talents of natural killers: NK cells in innate and adaptive immunity. EMBO Rep. 2009;10:1103-1110
17. Lappegård KT, Garred P, Jonasson L. et al. A vital role for complement in heart disease. Mol Immunol. 2014;61:126-134
18. Iltumur K, Karabulut A, Toprak G. et al. Complement activation in acute coronary syndromes. APMIS. 2005;113:167-174
19. Cusack MR, Marber MS, Lambiase PD. et al. Systemic inflammation in unstable angina is the result of myocardial necrosis. J Am Coll Cardiol. 2002;39:1917-1923
20. Leinoe E, Pachai A, Brandslund I. Complement activation reaches maximum during equilibrium between antigen and antibody in an in vitro model for thrombolysis with streptokinase. APMIS. 2000;10:685-688
21. Backström E, Kristensson K, Ljunggren HG. Activation of natural killer cells: underlying molecular mechanisms revealed. Scand J Immunol. 2004;60:14-22
22. Middleton D, Curran M, Maxwell L. Natural killer cells and their receptors. Transpl Immunol. 2002;10:147-64
23. Campbell KS, Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology. 2011;132:315-325
24. Raulet DH, Gasser S, Gowen BG. et al. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413-441
25. Zafirova B, Wensveen FM, Gulin M. et al. Regulation of immune cell function and differentiation by the NKG2D receptor. Cell Mol Life Sci. 2011;68:3519-3529
26. Koch J, Steinle A, Watzl C. et al. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013;34:182-191
27. Claus M, Meinke S, Bhat R. et al. Regulation of NK cell activity by 2B4, NTB-A and CRACC. Front Biosci. 2008;13:956-965
28. Whitman SC, Rateri DL, Szilvassy SJ. et al. Depletion of natural killer cell function decreases atherosclerosis in low-density lipoprotein receptor null mice. Arterioscler Thromb Vasc Biol. 2004;24:1049-1054
29. Li W, Lidebjer C, Yuan XM. et al. NK cell apoptosis in coronary artery disease: relation to oxidative stress. Atherosclerosis. 2008;199:65-72
30. Jonasson L, Backteman K, Ernerudh J. Loss of natural killer cell activity in patients with coronary artery disease. Atherosclerosis. 2005;183:316-321
31. Backteman K, Ernerudh J, Jonasson L. Natural killer (NK) cell deficit in coronary artery disease: no aberrations in phenotype but sustained reduction of NK cells is associated with low-grade inflammation. Clin Exp Immunol. 2014;175:104-112
32. Zehn D, King C, Bevan MJ. et al. TCR signaling requirements for activating T cells and for generating memory. Cell Mol Life Sci. 2012;69:1565-1575
33. Vanaki E, Ataei M, Sanati MH. et al. Expression patterns of Th1/Th2 transcription factors in patients with guttate psoriasis. Acta Microbiol Immunol Hung. 2013;60:163-174
34. Gao P, Zhou XY, Yashiro-Ohtani Y. et al. The unique target specificity of a nonpeptide chemokine receptor antagonist: selective blockade of two Th1 chemokine receptors CCR5 and CXCR3. J Leukoc Biol. 2003;73:273-280
35. Wan YY. GATA3: a master of many trades in immune regulation. Trends Immunol. 2014;35:233-242
36. Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277:2005-2007
37. Nagata K, Hirai H. The second PGD< sub> 2</sub> receptor CRTH2: structure, properties, and functions in leukocytes. Prostaglandins Leukot Essent Fatty Acids. 2003;69:169-177
38. Hastings WD, Anderson DE, Kassam N. et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009;39:2492-2501
39. Curtiss ML, Gorman JV, Businga TR. et al. Tim-1 regulates Th2 responses in an airway hypersensitivity model. Eur J Immunol. 2012;42:651-661
40. Gadhamsetty S, Marée AFM, Beltman JB. et al. A General Functional Response of Cytotoxic T lymphocyte -Mediated Killing of Target Cells. Biophys J. 2014;106:1780-1791
41. Keefe D, Shi L, Feske S. et al. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity. 2005;23:249-262
42. Berke G. The CTL's kiss of death. Cell. 1995;81:9-12
43. Kyaw T, Winship A, Tay C. et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. 2013;127:1028-1039
44. Zhou J, Dimayuga PC, Zhao X. et al. CD8 (+) CD25 (+) T cells reduce atherosclerosis in apoE (-/-) mice. Biochem Biophys Res Commun. 2014;443:864-870
45. Pike KA, Ratcliffe MJ. Cell surface immunoglobulin receptors in B cell development. Semin Immunol. 2002;14:351-358
46. Kurosaki T. Regulation of BCR signaling. Mol Immunol. 2011;48:1287-1291
47. Barrington RA, Schneider TJ, Pitcher LA. et al. Uncoupling CD21 and CD19 of the B-cell coreceptor. Proc Natl Acad Sci USA. 2009;106:14490-14495
48. Pospisil R, Silverman GJ, Marti GE. et al. CD5 is A potential selecting ligand for B-cell surface immunoglobulin: a possible role in maintenance and selective expansion of normal and malignant B cells. Leuk Lymphoma. 2000;36:353-365
49. Bergmann H, Yabas M, Short A. et al. B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8- dendritic cells require the intramembrane endopeptidase SPPL2A. J Exp Med. 2013;210:31-40
50. Thibult ML, Mamessier E, Gertner-Dardenne J. et al. PD-1 is a novel regulator of human B-cell activation. Int Immunol. 2013;25:129-137
Author contact
![]() Corresponding authors: Lemin Wang, Haoming Song, Yuqin Shen, Department of Cardiology, Tongji Hospital, Tongji University School of Medicine, 389 Xincun Rd, Putuo District, Shanghai 200065, China; Tel: 86 21 66111329, Fax: 86 21 66111329, E-mail: wangleminedu.cn; songhao-mingcom; sy-1963com.
Corresponding authors: Lemin Wang, Haoming Song, Yuqin Shen, Department of Cardiology, Tongji Hospital, Tongji University School of Medicine, 389 Xincun Rd, Putuo District, Shanghai 200065, China; Tel: 86 21 66111329, Fax: 86 21 66111329, E-mail: wangleminedu.cn; songhao-mingcom; sy-1963com.