Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(1):67-74. doi:10.7150/ijms.16907 This issue Cite
Research Paper
Specific networks of plasma acute phase reactants are associated with the severity of chronic obstructive pulmonary disease: a case-control study
Elena Arellano-Orden1 ![]() , Carmen Calero-Acuña1, 2,3, Juan Antonio Cordero1, María Abad-Arranz2, Verónica Sánchez-López1, Eduardo Márquez-Martín1, 2, Francisco Ortega-Ruiz1,2,3, José Luis López-Campos1,2,3
, Carmen Calero-Acuña1, 2,3, Juan Antonio Cordero1, María Abad-Arranz2, Verónica Sánchez-López1, Eduardo Márquez-Martín1, 2, Francisco Ortega-Ruiz1,2,3, José Luis López-Campos1,2,3
1. Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío/Universidad de Sevilla, Seville, Spain;
2. Unidad Médico-Quirúrgica de Enfermedades Respiratorias, Hospital Virgen del Rocío. Seville, Spain;
3. CIBER de Enfermedades Respiratorias (CIBERES), Instituto de Salud Carlos III, Madrid, Spain.
Received 2016-7-20; Accepted 2016-11-1; Published 2017-1-15
Abstract
Objectives. A detailed understanding of the intricate relationships between different acute phase reactants (APRs) in chronic obstructive pulmonary disease (COPD) can shed new light on its clinical course. In this case-control study, we sought to identify the interaction networks of a number of plasma APRs in COPD, with a special focus on their association with disease severity.
Methods. COPD cases and healthy smoking controls (3:1 ratio) were recruited in our outpatient pulmonary clinic. Cardiopulmonary exercise testing was used to rule out the presence of ischemic heart disease. All subjects were males as per protocol. Multiple plasma APRs - including α-2-macroglobulin, C-reactive protein (CRP), ferritin, fibrinogen, haptoglobin, procalcitonin (PCT), serum amyloid A (SAA), serum amyloid P, and tissue plasminogen activator (tPA) - were measured using commercial Acute Phase Bio-Plex Pro Assays and analyzed on the Bio-Plex manager software. Correlations between different APRs were investigated using a heat map. Network visualization and analyses were performed with the Cytoscape software platform.
Results. A total of 96 COPD cases and 33 controls were included in the study. Plasma A2M, CRP, and SAP levels were higher in COPD patients than in controls. Circulating concentrations of haptoglobin and tPA were found to increase in parallel with the severity of the disease. Increasing disease severity was associated with distinct intricate networks of APRs, which were especially evident in advanced stages.
Conclusions. We identified different networks of APRs in COPD, which were significantly associated with disease severity.
Keywords: acute phase reactants, chronic obstructive pulmonary disease
Introduction
Recent years have witnessed an increasing interest in the occurrence of systemic inflammation in COPD - which can explain, at least in part, its main extrapulmonary manifestations (1, 2). In general, the term systemic inflammation indicates an increase in plasma levels of various inflammatory proteins and acute phase reactants (APRs) belonging to different biological pathways. An elevation in circulating inflammatory markers may represent a potential therapeutic target (3, 4) for tackling the systemic burden of COPD (5), with several studies showing a significant adverse prognostic significance of increased APRs levels (6-8).
In all mammalian species, APRs are released from the liver to the systemic circulation mainly through the action of different proinflammatory cytokines (e.g., IL-6, IL-1 and TNF-α). Two distinct APRs classes can be distinguished based on the expression patterns elicited by cytokines on the liver. Class 1 APRs - which are mainly regulated by IL-1 or the combination of IL-1, IL-6, and glucocorticoids - include haptoglobin, C-reactive protein (CRP), serum amyloid A (SAA), α-1 acid glycoprotein (AGP), and hemopexin. Class 2 APRs - which are solely regulated by IL-6 and glucocorticoids - consist of fibrinogen, α-1 antichymotrypsin, and α-1 antitrypsin (9).
The most important research gaps that currently exist in the field of systemic inflammation in COPD include a) the exact source of APRs release, b) the potential interindividual variability of the inflammatory response, and c) how distinct inflammatory biomarkers can drive disease progression (4). Moreover, the observation that different APRs are not elevated in isolation supports the existence of intricate networks of different proinflammatory molecules that can fine-tune the systemic manifestations of COPD (10). Although there is evidence that combining information from different APRs may improve the prediction of progression in COPD, most studies to date have focused only on a limited number of different inflammatory biomarkers (10). Another major issue of the available investigations is the potential confounding effect of ischemic heart disease, which is a common comorbidity associated with systemic inflammation as well (11).
The identification of specific inflammatory signatures that may reflect disease severity in COPD is paramount for risk stratification and can shed new light on the disease course. To this aim, we designed the current study to perform an integrative analysis of different APRs. Specifically, the network visualization approach used in this report enabled us to obtain an overview of the complex relationships between different inflammatory markers, with a special focus on their association with disease severity. Owing to a potential confounding effect, female subjects and patients with ischemic heart disease were excluded from this study.
Methods
Study design and participants
This case-control study was conducted at the University Hospital Virgen del Rocío (Seville, Spain) between 2007 and 2010. Ethical approval was granted by the local Institutional Review Board (Comité de Ética e Investigación Clínica del Hospital Virgen del Rocío, Seville, Spain; approval act: 02/2006). Written informed consent was obtained from all participants. All analyses were performed in a cross-sectional fashion. COPD cases and healthy smoking controls (3:1 ratio) were recruited in our outpatient pulmonary clinic. Only male subjects were included to avoid the confounding effect of sex distribution. Inclusion criteria for COPD cases were as follows: 1) smokers and former-smokers with a diagnosis of COPD and a post-bronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio <0.7, 2) negative history of acute exacerbations in the previous three months, and 3) male sex. Smokers and former-smokers aged > 40 years with an FEV1/FVC ratio ≥ 0.7 were deemed eligible as controls. The measurement of exhaled carbon monoxide was used for confirming the smoking status in all participants. Exclusion criteria for both cases and controls included a previous history of ischemic heart disease, congestive heart failure, ventilator dependency, malignancies, hepatic cirrhosis, end-stage renal disease, rheumatologic disorders, tuberculosis, orthopedic conditions (that precluded or limited the performance in the walking and cardiopulmonary exercise tests), neurological or psychiatric illnesses that could interfere with the participation in the study, or any systemic inflammatory or infectious disease that could be associated with increased APRs levels. All participants underwent a cardiopulmonary exercise test coupling ECG with metabolic changes together with the clinical history and physical examination to rule out the presence of ischemic heart disease. In presence of positive results, the subject was excluded from the study and referred to the cardiology department for appropriate care.
Laboratory methods
Blood samples were drawn by venipuncture from each subject at rest. Samples were centrifuged at 3000 rpm for 5 min and stored at -80 °C until assayed. Plasma α-2-macroglobulin (A2M), C-reactive protein (CRP), ferritin, fibrinogen, haptoglobin, procalcitonin (PCT), serum amyloid A (SAA), serum amyloid P (SAP), and tissue plasminogen activator (tPA) concentrations were measured using commercially available Acute Phase Bio-Plex Pro Assays (BioRad Laboratories; Hercules, CA, USA) according to the manufacturer's protocol. The assay working ranges (defined by the ranges that extended from the lower to the upper limits of quantification) were as follows: 0.5-1875 ng/mL for A2M, 0.01-50 ng/mL for CRP, 3.05-50000 pg/mL for ferritin, 5-813 ng/mL for fibrinogen, 0.1-500 ng/mL for haptoglobin, 14-10000 pg/mL for PTC, 1-700 ng/mL for SAA, 0.1-200 ng/mL for SAP, and 28-5.000 pg/mL for tPA. All samples were blinded by a numerical code and laboratory personnel were unaware of the case-control status of each specimen. Measurements were performed in random order. All samples were analyzed in duplicate and the mean of the two measures was used for analysis. Plasma specimens (final volume: 50 μL) were diluted 100-fold for the measurements of A2M, CRP, ferritin, fibrinogen, and haptoglobin, whereas 10000-fold-diluted aliquots were used for quantifying PCT, SAA, SAP, and tPA. The analytical platform consisted of a 96-well plate-formatted, bead-based assay, with specific antibodies directed against the target proteins covalently coupled to the surfaces of the internally dyed bead sets. After a series of washing steps to remove unbound proteins, a biotinylated detection antibody specific for each epitope was added to the reaction. The beads were subsequently incubated with a reporter streptavidin-phycoerythrin (SA-PE) conjugate, and fluorescence of the bound SA-PE was measured through the specific array reader.
Data acquisition and analysis
All analytical data were acquired using the Bio-Plex platform (Bio-Rad Laboratories, Hercules, CA, USA), consisting of a suspension array system, a dual laser, and a flow-based microplated reader. The laser and associated optics are designed to detect the internal fluorescence of the individual dyed beads. The fluorescent signal on the bead surface is proportional to the quantity of target protein in the biological sample. All of the data were analyzed on the Bio-Plex manager software.
Statistical analysis
All calculations were conducted in the computing environment R (version 3.3.0; R Foundation for Statistical Computing, Vienna, Austria). Data pre-processing was performed by log-transformation and removal of out-of-range outliers. For each APR, we also considered as outliers all of the measures that fell outside the interval comprised between the first quartile minus two times the interquartile range (IQR) and the third quartile plus two times the IQR. Because data had a skewed distribution according to the Shapiro-Wilk test (p-value < 0.05), only non-parametric tests were applied to compare distributions. A network was computed based on the pairwise correlations between APRs. Edges were present when the calculation of the Spearman's correlation coefficient identified a statistically significant association (p-value < 0.05). Edge width represented the absolute correlation coefficient value, whereas color indicated the presence of a negative (black) or a positive (grey) association. Node size was proportional to the APR concentration. The heatmaps denoted the similarities in terms of biomarker profiles both in COPD patients and in control individuals. Spearman's correlation was considered as a similarity measure and numbers in each cell were the p-values for every correlation between different APRs. Because age was found to differ significantly between COPD patients and control individuals (Mann-Whitney U test), the effect of age on APRs levels was further tested using linear models. Differences between COPD patients and healthy controls, as well as across different disease stages, were calculated with the Mann-Whitney U test.
Results
A total of 96 COPD cases and 33 controls were included in the study. The general characteristics of the study participants are summarized in Table 1. The distribution of COPD stages was as follows: GOLD I, 23 patients (24%); GOLD II, 30 patients (31.2%); GOLD III, 28 patients (29.2%), and GOLD IV, 15 patients (15.6%).
General characteristics of the study participants.
| Control subjects (n=33) | COPD patients (n=96) | p value* | |
|---|---|---|---|
| Males (n) | 33 (100%) | 96 (100%) | NS |
| Age (years) | 58 (10) | 67 (8) | <0.001 |
| Tobacco history (pack-years) | 46.9 (27.8) | 71.9 (76.6) | 0.007 |
| Body mass index (kg/m2) | 28.78 (5) | 28.27 (4.8) | NS |
| Charlson-age index | 2.24 (1.6) | 3.87 (1.2) | <0.001 |
| FVC (%) | 91.59 (13.6) | 91.96 (20.9) | NS |
| FEV1 (%) | 90.26 (13.1) | 59.15 (22.8) | <0.001 |
Concentrations of acute phase reactants
There were not significant differences in the levels of A2M, CRP, ferritin, fibrinogen, haptoglobin, SAA and SAP between COPD patients and control subjects (Figure 1). However, plasma PCT and tPA levels were significantly higher in controls than in COPD patients (p=0.0211 and p=0.0434, respectively). Plasma levels of CRP, haptoglobin, PCT, and tPA were found to increase in parallel with disease severity (data not shown).
Levels of A2M (panel A), CRP (panel B), Ferritin (panel C), Fibrinogen (panel D), Haptoglobin (panel E), PCT (panel F), SAA (panel G) and tPA (panel H) in COPD cases and healthy smoking controls.

Associations between different biomarkers and identification of networks
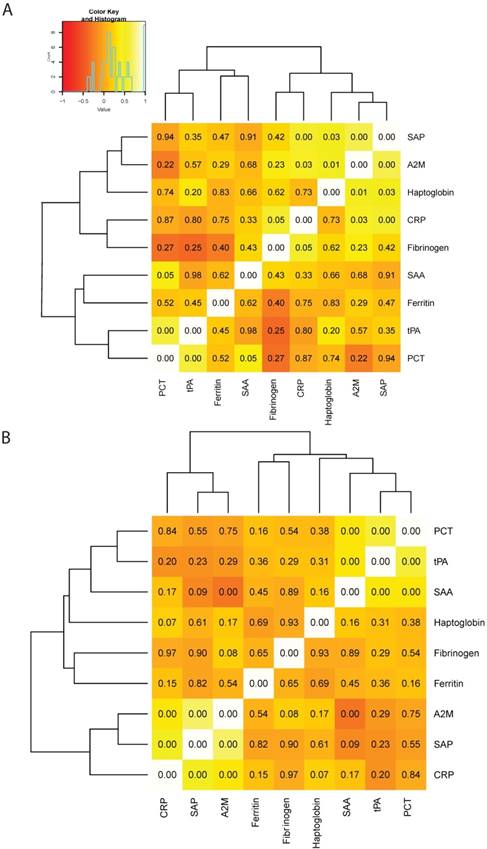
Correlations between different APRs were summarized in a heat map (Figure 2). The most robust correlations in the control group were those between A2M and SAP (r=0.835, p<0.001), PCT and SAA (r=0.708, p=0.05), as well as PCT and tPA (r=0.877, p<0.001). Conversely, the most marked correlation in COPD patients was that between SAP and A2M (r=0.713, p<0.001). Consequently, we identified a cluster formed by SAP, A2M, haptoglobin, and CRP in controls (Figure 2A). Two clusters were evident in COPD patients, the first being between PCT, tPA, and SAA and the second between CRP, SAP, and A2M (Figure 2B).
Heat map depicting the correlations between di_erent in_ammatory biomarkers in healthy smoking controls (panel A) and COPD cases (panel B). The intensity of the color re_ects the correlation coe_cient, whereas the number in each square indicates the p value.
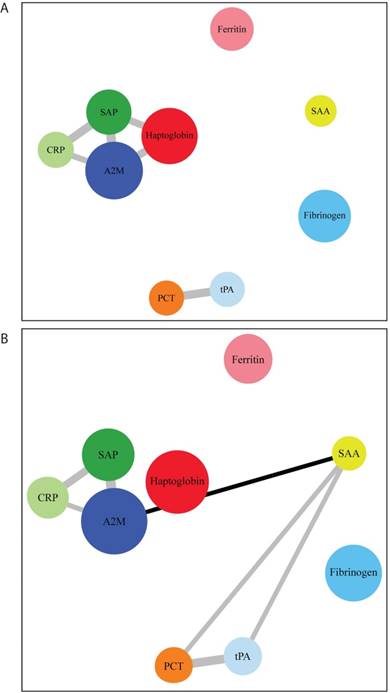
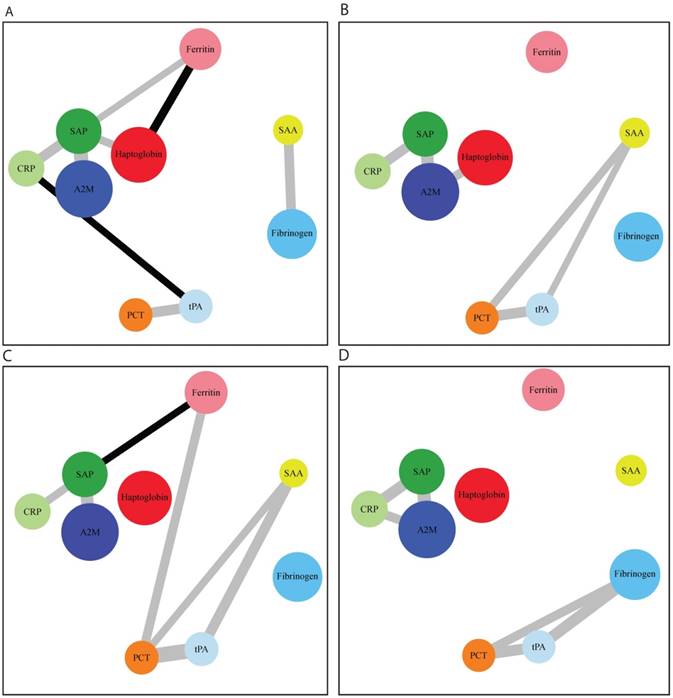
After network modelling (Figure 3), we were able to identify distinct networks of inflammatory biomarkers according to the presence and severity of COPD. In control subjects, there was a relation among CRP, SAP, haptoglobin, and A2M, as well as another association between tPA and PCT. In COPD, we found an additional correlation of SAA with all of these networks. We then analyzed the networks in relation to GOLD functional stages (Figure 4). GOLD I was characterized by a nonspecific increase in all of the assayed APRs. However, specific networks of inflammatory markers were identified as the severity of the disease increased. The intermediate disease stages (GOLD II and III) were characterized by an increased extent of the associations between different APRs. Interestingly, advanced disease stages (GOLD III-IV) showed inflammatory networks in which haptoglobin was independent from the cluster formed by SAP, CRP, and A2M. Finally, stage IV was characterized by a new cluster consisting of fibrinogen, PCT, and tPA.
Distinct networks of acute phase reactants in healthy smoking controls (panel A) and COPD patients (panel B). Node colors indicate the type of correlation (grey and black denote positive and negative correlations, respectively), whereas node size is proportional to the extent of correlation.
Distinct networks of acute phase reactants according to the severity of COPD. Panel A: patients with GOLD I COPD; panel B: patients with GOLD II COPD; panel C: patients with GOLD III COPD; panel D: patients with GOLD IV COPD. Node colors indicate the type of correlation (grey and black denote positive and negative correlations, respectively), whereas node size is proportional to the extent of correlation.
Discussion
In this study conducted in male COPD patients without comorbid ischemic heart disease, we performed a comprehensive assessment of APRs by network analysis. As expected, APRs were found to be increased in COPD. More importantly, we demonstrated that these biomarkers were reciprocally interrelated, being associated in complex interactive networks that were related to the severity of COPD.
APRs represent a super-family of different molecules whose circulating concentrations change by at least 25% in response to acute adverse stimuli. Specifically, positive acute-phase proteins show a significant increase during inflammation, whereas negative acute-phase reactants are characterized by corresponding reductions (12). Changes in APRs concentrations are generally believed to reflect their synthesis rates in the hepatocytes. However, the magnitude of inflammation-related APRs changes varies widely between different molecules. For example, levels of ceruloplasmin and several components of the complement system may increase by approximately 50 percent during inflammation, whereas up to 1000-fold increases have been reported for CRP and SAA (13).
In this study, we performed a number of APRs measurements to analyze the inflammatory networks in COPD. We uncovered intricate relationships between different inflammatory parameters in relation to the severity of the disease. Moreover, we confirmed that COPD patients free of ischemic heart disease are characterized by a significant elevation in APRs compared with healthy smoking controls. Of the different inflammatory markers, we observed a significant stepwise elevation in tPA and haptoglobin levels with increasing disease severity.
Our results may pave the way for longitudinal investigations on the prognostic significance of the inflammatory signatures identified in our study using independent and larger sample sets (14). Additionally, the potential impact of the COPD clinical phenotype (15) on the inflammatory networks needs to be examined. The question as to whether the markers or signatures herein identified could reflect subtle interindividual differences in the inflammatory expression of COPD remains open. However, an important strength inherent in our study is the careful exclusion of patients with ischemic heart disease through cardiopulmonary exercise testing. Because cardiovascular disorders are closely intertwined with COPD and can act as a major confounder, we deemed all patients with a positive exercise test not eligible for inclusion. The relationship between COPD and ischemic heart disease is well-known (16) and both conditions share common risk factors, including age and tobacco smoke (17). In turn, the systemic inflammatory reaction occurring in COPD (18) may increase the risk of developing vascular manifestations (19, 20).
Published data on the concomitant changes of different inflammatory biomarkers in COPD are scarce. In the Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE) study (7), the authors measured a total of six inflammatory biomarkers (white blood cell count, CRP, interleukin [IL]-6, IL-8, fibrinogen, and tumor necrosis factor [TNF]-α) in three groups of subjects (1755 COPD patients, 297 smokers with normal results on spirometry, and 202 non-smokers) over a 3-year follow-up. A persistent systemic inflammatory load was observed in a significant proportion of the study patients. Moreover, specific interplays between different biomarkers were noted in COPD patients compared with healthy smokers (i.e., increased white blood cell count, CRP, IL-6, and fibrinogen accompanied by a decreased expression of IL-8 and TNF-α).
One of the most innovative aspects of our study is the identification of specific networks of plasma APRs as significantly associated with the severity of COPD. We identified a strong association between CRP and SAP, which act as opsonins. Conversely, we did not find an association between SAA and CRP. Although these molecules share similar secretory stimuli (13), CRP mainly activates the complement system whereas SAA acts predominantly on leucocytes. Interestingly, we demonstrate for the first time that fibrinogen is not part of the inflammatory networks that characterize mild-to-moderate COPD. Fibrinogen is a class II acute phase reactant and is solely regulated by IL-6 and glucocorticoids (9). Although it has been suggested that fibrinogen may predict prognosis (6) and be part of a specific inflammatory phenotype in COPD (10), it is worth noting that previous studies did not exclude patients with cardiovascular disorders, in whom fibrinogen levels are notoriously increased (24). Because this potential confounder has been removed from our study, we believe that future research should reevaluate the potential role of fibrinogen in COPD. However, we have found an association between fibrinogen and tPA in advanced COPD (GOLD IV); both molecules participate in the coagulation cascade. Fibrinogen is implicated in clot formation whereas tPA plays a role in its dissolution, with both of these phenomena being present in patients with advanced disease.
Among the inflammatory networks of COPD, network analysis revealed a prominent association between PCT and tPA. PCT is the precursor of calcitonin, whose serum levels have been studied in patients with respiratory infections as a marker for guiding antibiotic therapy during exacerbations (25). Although a recent report demonstrated a correlation of salivary PCT levels with both breathing scores and sputum features in COPD (26), to our knowledge the potential impact of plasma PCT in patients with stable disease has not been previously investigated. However, its potential role as a biomarker is worth of investigation in future studies.
The present study must be evaluated in the light of several limitations. In the current report, we limited our analysis to male subjects. Different studies have shown that sex may have a significant impact in the clinical presentation of COPD from both the clinical (21) and inflammatory (22, 23) standpoints. Further research is needed to investigate whether our current findings may be applied to females as well. The severity of COPD was evaluated with FEV1 alone. However, we know that the evaluation of severity can be more complex and comprehensive. In this regard, future studies can evaluate the impact of the inflammatory load on different clinical features. Finally, other non-cardiac comorbidities may also influence the results, since several chronic diseases have been associated with elevated systemic biomarkers, although we did not find any association between biomarkers and comorbidities.
In summary, we performed an integrative statistical analysis of different inflammatory markers in COPD after the exclusion of patients with ischemic heart disease. Importantly, we were able to identify different networks of APRs which were significantly associated with COPD severity. Pending external validation, the markers or signatures herein identified could be helpful for patient monitoring, stratification in clinical trials, or personalizing existing or upcoming anti-inflammatory therapies.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Garcia-Rio F, Miravitlles M, Soriano JB, Munoz L, Duran-Tauleria E, Sanchez G. et al. Systemic inflammation in chronic obstructive pulmonary disease: a population-based study. Respiratory research. 2010;11:63
2. Pinto-Plata V, Toso J, Lee K, Park D, Bilello J, Mullerova H. et al. Profiling serum biomarkers in patients with COPD: associations with clinical parameters. Thorax. 2007;62(7):595-601
3. Janssen DJ, Mullerova H, Agusti A, Yates JC, Tal-Singer R, Rennard SI. et al. Persistent systemic inflammation and symptoms of depression among patients with COPD in the ECLIPSE cohort. Respiratory medicine. 2014;108(11):1647-54
4. Faner R, Tal-Singer R, Riley JH, Celli B, Vestbo J, MacNee W. et al. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax. 2014;69(7):666-72
5. Pinto-Plata VM, Mullerova H, Toso JF, Feudjo-Tepie M, Soriano JB, Vessey RS. et al. C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax. 2006;61(1):23-8
6. Duvoix A, Dickens J, Haq I, Mannino D, Miller B, Tal-Singer R. et al. Blood fibrinogen as a biomarker of chronic obstructive pulmonary disease. Thorax. 2013;68(7):670-6
7. Thorleifsson SJ, Margretardottir OB, Gudmundsson G, Olafsson I, Benediktsdottir B, Janson C. et al. Chronic airflow obstruction and markers of systemic inflammation: results from the BOLD study in Iceland. Respiratory medicine. 2009;103(10):1548-53
8. Dahl M, Vestbo J, Lange P, Bojesen SE, Tybjaerg-Hansen A, Nordestgaard BG. C-reactive protein as a predictor of prognosis in chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 2007;175(3):250-5
9. Lyoumi S, Tamion F, Petit J, Dechelotte P, Dauguet C, Scotte M. et al. Induction and modulation of acute-phase response by protein malnutrition in rats: comparative effect of systemic and localized inflammation on interleukin-6 and acute-phase protein synthesis. The Journal of nutrition. 1998;128(2):166-74
10. Agusti A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, Miller BE. et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PloS one. 2012;7(5):e37483
11. Stolz D, Meyer A, Rakic J, Boeck L, Scherr A, Tamm M. Mortality risk prediction in COPD by a prognostic biomarker panel. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 2014;44(6):1557-70
12. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. The New England journal of medicine. 1999;340(6):448-54
13. Steel DM, Whitehead AS. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol Today. 1994;15(2):81-8
14. Boue S, Fields B, Hoeng J, Park J, Peitsch MC, Schlage WK. et al. Enhancement of COPD biological networks using a web-based collaboration interface. F1000Res. 2015;4:32
15. Lopez-Campos JL, Bustamante V, Munoz X, Barreiro E. Moving towards patient-centered medicine for COPD management: multidimensional approaches versus phenotype-based medicine-a critical view. Copd. 2014;11(5):591-602
16. Williams MC, Murchison JT, Edwards LD, Agusti A, Bakke P, Calverley PM. et al. Coronary artery calcification is increased in patients with COPD and associated with increased morbidity and mortality. Thorax. 2014;69(8):718-23
17. Sinha SS, Gurm HS. The double jeopardy of chronic obstructive pulmonary disease and myocardial infarction. Open Heart. 2014;1(1):e000010
18. Mullerova H, Agusti A, Erqou S, Mapel DW. Cardiovascular comorbidity in COPD: systematic literature review. Chest. 2013;144(4):1163-78
19. Engstrom G, Lind P, Hedblad B, Wollmer P, Stavenow L, Janzon L. et al. Lung function and cardiovascular risk: relationship with inflammation-sensitive plasma proteins. Circulation. 2002;106(20):2555-60
20. Sin DD, Man SF. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003;107(11):1514-9
21. Roche N, Deslee G, Caillaud D, Brinchault G, Court-Fortune I, Nesme-Meyer P. et al. Impact of gender on COPD expression in a real-life cohort. Respiratory research. 2014;15:20
22. Maury J, Gouzi F, De Rigal P, Heraud N, Pincemail J, Molinari N. et al. Heterogeneity of Systemic Oxidative Stress Profiles in COPD: A Potential Role of Gender. Oxid Med Cell Longev. 2015;2015:201843
23. Faner R, Gonzalez N, Cruz T, Kalko SG, Agusti A. Systemic inflammatory response to smoking in chronic obstructive pulmonary disease: evidence of a gender effect. PloS one. 2014;9(5):e97491
24. Danesh J, Lewington S, Thompson SG, Lowe GD, Collins R, Kostis JB. et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA: the journal of the American Medical Association. 2005;294(14):1799-809
25. Tokman S, Schuetz P, Bent S. Procalcitonin-guided antibiotic therapy for chronic obstructive pulmonary disease exacerbations. Expert review of anti-infective therapy. 2011;9(6):727-35
26. Patel N, Belcher J, Thorpe G, Forsyth NR, Spiteri MA. Measurement of C-reactive protein, procalcitonin and neutrophil elastase in saliva of COPD patients and healthy controls: correlation to self-reported wellbeing parameters. Respiratory research. 2015;16:62
Author contact
![]() Corresponding author: Elena Arellano-Orden, Instituto de Biomedicina de Sevilla (IBiS), Avda. Manuel Siurot, s/n.41013 Seville, Spain. E-mail: marellano-ibises.
Corresponding author: Elena Arellano-Orden, Instituto de Biomedicina de Sevilla (IBiS), Avda. Manuel Siurot, s/n.41013 Seville, Spain. E-mail: marellano-ibises.