Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(7):517-523. doi:10.7150/ijms.15507 This issue Cite
Research Paper
Effects of LG268 on Cell Proliferation and Apoptosis of NB4 Cells
Ting Xu1,2, Liang Zhong2, Liu-Gen Gan1,2, Chun-Lan Xiao1,2, Zhi-Ling Shan2, Rong Yang2, Hao Song2, Liu Li2, Bei-Zhong Liu1,2 ![]()
1. Central Laboratory of Yong-chuan Hospital, Chongqing Medical University, Chongqing, 402160, China
2. Key Laboratory of Laboratory Medical Diagnostics, Ministry of Education, Department of Laboratory Medicine, Chongqing Medical University, Chongqing, 400016, China
Received 2016-3-11; Accepted 2016-6-12; Published 2016-6-29
Abstract
Aims: To investigate the effect of LG100268 (LG268) on cell proliferation and apoptosis in NB4 cells.
Methods: NB4 cells were treated with LG268 for 24 h or 48 h. The effect of LG268 on cell proliferation was assessed by the CCK-8 assay and colony-forming assay. Apoptosis and cell cycle were evaluated by flow cytometry. The protein expression levels of Survivin, PARP, c-Myc, cyclin D1, ERK, p-ERK, p38 MAPK, and p- p38 MAPK were detected by western blot.
Results: We found that LG268 inhibited the proliferation of NB4 cells in a dose-dependent manner. Flow cytometry analysis showed that LG268 accelerated apoptosis in NB4 cells in a time- dependent manner and that LG268 treatment led to cell cycle arrest at G0/G1 phase. Moreover, LG268 significantly decreased the protein levels of Survivin, c-Myc, and cyclinD1. Cleaved PARP was observed in the LG268 treatment group but not in the control group. In addition, LG268 increased the phosphorylation level of p38 MAPK and decreased the phosphorylation level of ERK.
Conclusions: LG268 inhibited cell proliferation and promoted cell apoptosis in NB4 cells.
Keywords: LG268, NB4 cells, proliferation, apoptosis, ERK, p38 MAPK
Introduction
Acute promyelocytic leukemia (APL) accounts for approximately 10-15% of acute myeloid leukemia (AML) [1]. APL is characterized by a chromosomal t(15;17) translocation, which forms the oncogenic PML-RARA fusion protein. Clinically, all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) have been useful for curing the great majority of patients with APL [2, 3]. However, 10-30% of APL patients are not sensitive to ATRA and ATO [4]. Thus, APL remains a challenging disease to treat and urgently requires new therapeutic strategies.
Retinoid X receptors (RXR) are common heterodimerization partners for many nuclear receptors (NRs), and therefore control a variety of physiological processes. The loss of RXRα is lethal during fetal development due to hypoplasia of the myocardium [5]. Several RXR agonists are reportedly effective in suppressing the progress of multiple diseases, including hepatocellular carcinoma [6], metabolic syndrome [7], neurodegeneration [8], Parkinson's disease [9], and breast cancer [10]. Moreover, some researchers have reported RXR involvement in leukemia treatment, and it regulated genes involved in myeloid differentiation [11-15]. LG268, the pure RXR agonist, is specific for RXR, and shows no appreciable binding to retinoic acid receptors [16]. LG268 participates in many cancer prevention and treatment processes. For example, rexinoid LG268 effectively prevented the development of both malignant and premalignant mammary lesions in MMTV-ErbB2 mice [17] and suppressed lung carcinogenesis in A/J mice [18]. Furthermore, it was reported that LG268 increased apoptosis and necrosis in the inner cell mass (undifferentiated cells) of bovine embryos, whereas ATRA had no effect [19]. Indeed, because of its highly specific binding to RXR, LG268 has been reported to execute an apoptotic program in rats without undesirable toxicity [20]. However, studies of the effect LG268 on APL are rarely.
In this study, we used NB4 cells, an APL cell line, to examine the effects of LG268 on cell biological properties. We found that LG268 affected NB4 cell proliferation and apoptosis. Our observations suggest that LG268 could provide a new therapeutic approach for the treatment of APL.
Materials and Methods
Materials
LG100268 was purchased from Sigma (St Louis, MO, USA). The antibodiy against p-p38 MAPK was purchased from Millipore (USA). Antibodies against p38 MAPK, PARP, ERK, p-ERK, and c-Myc were from Cell Signaling Technology (USA), Antibodies against Survivin and cyclinD1 were from Wanleibio (China). Goat anti-rabbit antibody, goat anti-mouse antibody and anti-β-actin antibody were purchased from Zhongshan Goldenbrige Biotechnology (China).
Cell culture
NB4 cells (Institutes for Biological Sciences, Shanghai, China) were cultured in RPMI 1640 (Gibco Life Technologies, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco Life Technologies) supplemented with penicillin (100 units/mL) and streptomycin (100 mg/mL) at 37 °C in an environment containing 5% CO2. The complete medium was refreshed daily.
CCK-8 assay
The cells were seeded into 96‑well plates (1×104cells/well), and 10 μL CCK‑8 (7Sea Cell Counting Kit; Sevenseas Futai Biotechnology Co., Ltd., Shanghai, China) was added to each well. After incubating 2 h, the absorbance of each well was measured at 450 nm using a spectrophotometer (Bio‑Rad Laboratories, Inc). The experiment was repeated three times.
Colony forming assay
NB4 cells were exposed to LG268 or DMSO for 48h. Then, cells were plated in 24-well plates in methylcellulose medium in triplicate. The numbers of colonies were determined following incubation for 14 days at 37 °C and 5 % CO2.
Flow cytometric assay
Cells were washed using PBS. And the cell pellets were resuspended and stained with annexin V-FITC and propidium iodide (PI) (Sigma‑Aldrich). The rate of cell apoptosis was analyzed using a FACsorter (BD Biosciences, San Jose, CA, USA) after incubating 15 min at room temperature. For cell cycle detection, cells were fixed with precooled 70% ethanol overnight at -20°C. After centrifugation, the cells were resuspended with RNase solution in a 37°C water bath for 30 min. Then propidium iodide staining solution was added and incubated for 30 min in the dark at room temperature. The cell cycle distribution was determined using a FACsorter.
Western blot analysis
Cells were lysed in radioimmunoprecipitation solution containing protease inhibitor phenylmethanesulfonyl fluoride (PMSF), phosphatase inhibitor NaF and Na3VO3. The protein concentration was quantified using the BCA method. Equal amounts of extracted total protein (30 μg) were separated by 10% sodium dodecyl sulfate‑polyacrylamide gel electrophoresis, and subsequently transferred onto polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA). The membrane was blocked with 5% skim milk for 2 h at room temperature and then incubated with the primary antibodies (1:500) overnight at 4°C.The membranes were then incubated with goat anti‑rabbit and goat anti‑mouse secondary antibodies (1:4000) for 1 h at 37°C. After washing with Tris‑buffered saline containing Tween‑20 (TBST), the immunoreactive complexes were visualized using an enhanced chemiluminescence system. β‑actin was used as the internal positive control.
Statistical analysis
Statistical analyses were performed using SPSS 17.0 software (SPSS Inc., Chicago, IL, USA). Experimental results are presented as the mean ± standard deviation. An independent samples t‑test was used to compare the results of two groups. Statistical analysis of western blot results was performed using analysis of variance (ANOVA). P < 0.05 was considered a statistically significant. Each experiment was repeated at least three times.
Results
Growth effects of LG268
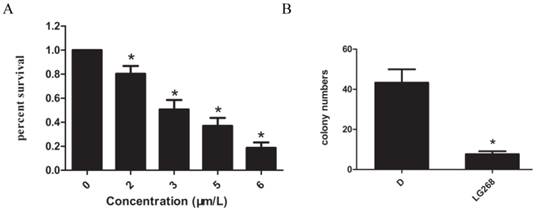
The effect of LG268 on the viability of NB4 cells was evaluated by CCK-8 assay. Treatment with different concentrations (0-6 μM) of LG268 for 48 h resulted in a dose-dependent reduction in cellular viability (Fig. 1A). Based on these observations, 2 μM LG268 was chosen for the cloning assay. Exposure to 2 μM LG268 significantly inhibited the colony-forming capacity of NB4 cells compared with DMSO treatment (P < 0.05) (Fig. 1B).
LG268 inhibited the proliferation of NB4 cells. (A) Cytotoxicity was detected by CCK-8 assay. Data are expressed as means ± SD. (B) The in vitro colony-forming ability of NB4 cells was examined in the absence or presence of 2 μM LG268. *P < 0.05.
Cell cycle analysis
The cytotoxic effects of LG268 on NB4 cells were further confirmed by cell cycle analysis. Compared to the negative control, LG268 dramatically increased the percentage of cells in the G0/G1 phase from 47.26% to 80.44% (Fig. 2A).
LG268 induced G0/G1 arrest in NB4 cells. NB4 cells were treated with 3 μM LG268 for 48 h. (A) Cells were harvested for analysis of cell cycle distribution by flow cytometry. (B) Effect of LG268 on the expression of c-Myc and cyclin D1 determined by western blot analysis. Quantitative analysis was performed by measuring the relative expression level of c-Myc and cyclin D1 to that of β-Actin. Data are expressed as means ± SD. *P < 0.05.
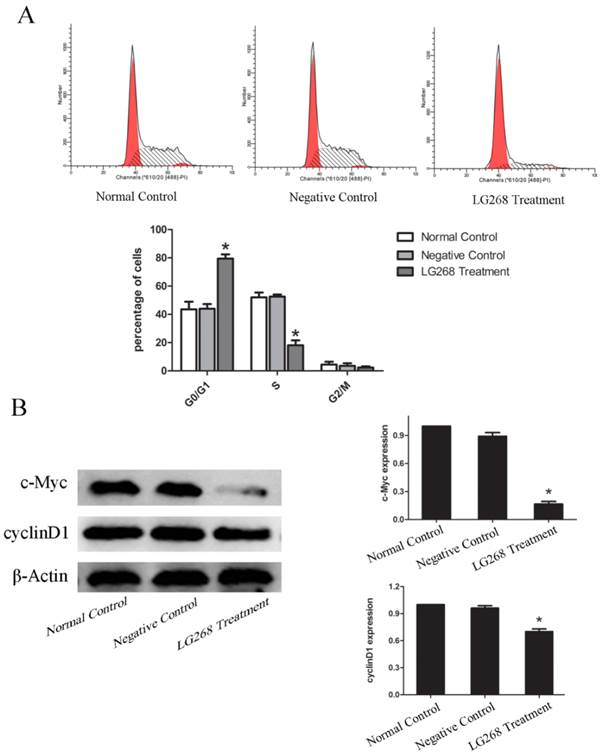
Next, we measured the expression levels of several cell -cycle-related proteins by western blotting. LG268 treatment greatly decreased the protein levels of cyclin D1 and c-Myc (Fig. 2B). These data provided strong evidence that LG268 inhibits proliferation of NB4 cells by inducing G0/G1 phase cell-cycle arrest.
Effect of LG268 on apoptosis of NB4 cells
To test whether LG268 affected NB4 cell apoptosis, we evaluated the intensity of apoptosis by Annexin V-FITC and PI staining and flow cytometry. Cellular apoptosis was significantly increased in the LG268-treated groups relative to the negative control groups (Fig. 3A). Treatment of NB4 cells with LG268 for 48 h induced higher levels of apoptosis than treatment for 24 h. These results indicated that LG268 could promote apoptosis in NB4 cells in a time-dependent manner.
LG268 induced apoptosis of NB4 cells. (A) Cells were treated with LG268 for 24 h or 48 h, and apoptosis was analyzed by flow cytometry using double staining with FITC-labeled annexin-V and propidium iodide. Cells undergoing early apoptosis are Annexin V-FITC+/PI- , whereas cells undergoing late apoptosis are Annexin V-FITC+/PI+. The percentages of late and early apoptotic cells were summed to give the total number of apoptotic cells. (B) Cells were treated with LG268 for 48 h, and the effect of LG268 on the expression of cleaved-PARP and Survivin were determined by western blot analysis. Quantitative analysis was performed by measuring the relative expression level of cleaved-PARP and Survivin to β-Actin. Data are expressed as means ± SD. *P < 0.05.
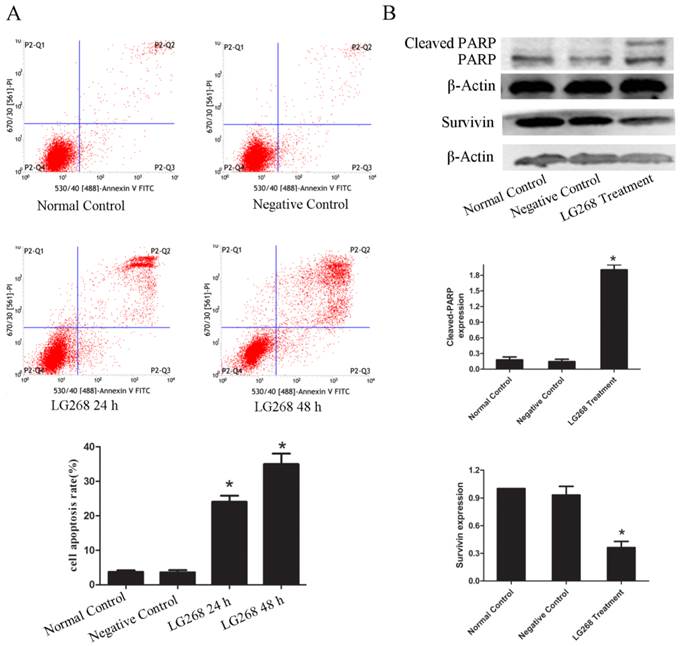
To further assess the apoptotic effects of LG268 treatment on NB4 cells, we examined the expression levels of the apoptosis regulatory proteins Survivin and cleaved poly-(adenosine diphosphate-ribose) polymerase (PARP). The level of Survivin protein was notably downregulated with LG268 treatment (Fig. 3B). Consistently, cleaved PARP was observed in the LG268 treatment group but not in the control group (Fig. 3B).
Effect of LG268 on multiple signaling molecules in NB4 cells
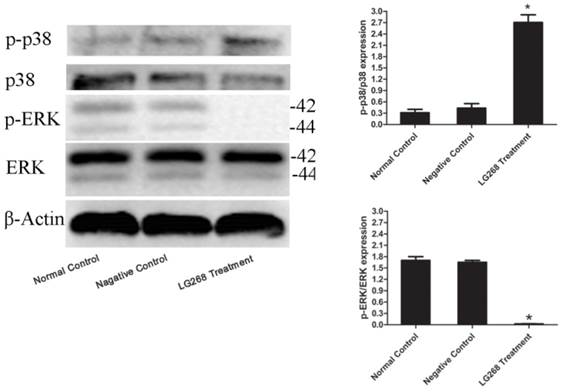
To investigate the molecular mechanism of LG268 on NB4 cells, we measured the phosphorylation levels of ERK and p38 MAPK by immunoblot analysis of whole cell lysates extracted from LG268-treated cells. Fig. 4 shows that 3 μM LG268 significantly decreased the level of phosphorylated ERK, whereas it increased the phosphorylation level of p38 MAPK.
LG268 affected multiple signaling molecules. Western blot analysis was used to measure the expressions of phosphorylated ERK and p38 MAPK. Quantitative analysis was performed by measuring the relative expression level of p-p38 to p38 or p-ERK to ERK. Data are expressed as means ± SD. *P < 0.05.
Discussion
APL is a malignancy that is highly curable using targeted therapies [21]. ATRA and ATO lead to complete remission in most patients with APL, but a large proportion of patients eventually experience a relapse [22-24]. Therefore, novel agents are necessary to improve the outcomes for patients with APL. This study investigated the effects of LG268, the pure RXR agonist, on an APL cell line. Our CCK-8 assay showed that, NB4 cell proliferation was inhibited by 3 μM LG268 in a dose-dependent manner. Because treatment with 3 µM LG268 resulted in very few colonies, we applied 2 µM LG268 for the colony-forming assay, which confirmed that LG268 could suppress NB4 cell proliferation.
As a general transcription factor, the c-Myc oncoprotein regulates the expression of a large number of genes involved in the cell cycle, apoptosis, and differentiation [25-26]. In normal cells, inhibition of c-Myc invariably results in a G0/G1 cell cycle arrest [27]. In contrast, tumor cells expressing c-Myc-shRNAs display several distinct types of cell cycle arrest [28]. Cyclin D1 is closely associated with the proliferation of cancer cells, and it has an important role in the G1/S-phase transition, which may promote the occurrence of tumors [29-30]. In this study, we found that LG268 treatment remarkably reduced the expression of c-Myc and cyclin D1, which may subsequently arrest the cell cycle at the G0/G1 phase. These data provided strong evidence that LG268 inhibits proliferation of NB4 cells by inducing G0/G1-phase cell-cycle arrest. Apoptosis can occur before the first mitosis (early apoptosis) or as the last step of mitotic catastrophe (late apoptosis) [31]. In the early stage, changes in the cellular membrane leading to phosphatidylserine eversion, and chromatin condensation, DNA breaks, and apoptotic body formation appear in the mid-late stage. LG268 mostly promoted late apoptosis of NB4 cells, as detected by flow cytometry. Survivin is a critical inhibitor of apoptosis-inducing proteins that is up-regulated in many cancers and considered a critical target for cancer therapy [32]. Previous studies associated high expression of Survivin with aggressive disease status and predicted poor clinical outcomes in AML [33-36]. We found that LG268 inhibited the expression of Survivin in NB4 cells, which suggests that Survivin could be the therapeutic target of LG268 for APL. Accordingly, cleavage of the caspase-3 target, PARP, was only observed in LG268-treated NB4 cells. PARP activation inhibits PI3K/Akt pathway and consequently inhibits cell apoptosis [37]; thus, increased expression levels of cleaved PARP may promote cell apoptosis.
Our study demonstrated that the level of phosphorylated ERK1/2 was reduced after LG268 treatment, which suggests that LG268 blocked MEK/ERK signaling in APL cells. Previous studies indicated that blast cells from most cases of AML, including APL, showed constitutive activation of ERK 1/2 [38]. The MEK/ERK pathway possesses anti-apoptotic functions and has profound effects on the growth regulation of malignant hematopoietic cells [39-40]. Moreover, it was reported that the inhibition of MEK activation reduced PML/RARa expression in both NB4 cells and primary APL blast cells [41]. These findings indicated that MEK/ERK signaling might play a role in LG268-induced cytotoxicity and apoptosis. p38 MAPK regulates cell proliferation, differentiation, apoptosis, and senescence [42-43], and the levels of phosphorylated p38 are lower in CD34+/CD38- AML cells than in normal hematopoietic stem cells [44]. In this study, LG268 induced the phosphorylation of p38 MAPK, which suggested that the observed apoptotic changes might depend on p38 MAPK signaling.
Previous studies reported that LG268 induces myeloid maturation of AML cell lines [45]; however, we failed to detect any changes in the myelomonocytic markers CD11b and CEBPβ between the control group and LG268 treatment group (data not shown). This differential outcome might arise from the different concentration or treatment duration employed, different cell status or other experimental differences.
In conclusion, the present study indicates that LG268 could inhibit growth of NB4 cells by inducing apoptosis and cell -cycle arrest. LG268 treatment inhibited the expression of c-Myc, cyclin D1, and Survivin, and resulted in PARP cleavage in NB4 cells. Given the increased phosphorylation level of p38 MAPK and the decreased phosphorylation levels of ERK, we suggest that the MEK/ERK and p38 MAPK pathways may be involved in the pathogenesis of APL through their effects on proliferation and apoptosis. Thus, LG268 might be a useful candidate for chemotherapy against APL.
Acknowledgements
The present study was supported by a grant from the National Natural Science Foundation of China (grant no. 81171658) and the Natural Science Foundation Project of CQ CSTC (grant no. 2011BA5037).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93:3167-215
2. Hassani S, Ghaffari SH, Zaker F. et al. Azidothymidine hinders arsenic trioxide-induced apoptosis in acute promyelocytic leukemia cells by induction of p21 and attenuation of G2/M arrest. Ann Hematol. 2013;92:1207-20
3. Grimwade D, Enver T. Acute promyelocytic leukemia: where does it stem from? Leukemia. 2004;18:375-84
4. Kawasaki K, Akaike H, Miyauchi A. et al. Sivelestat relieves respiratory distress refractory to dexamethasone in all-trans retinoic acid syndrome: a report of two cases. Eur J Haematol. 2006;77:448-52
5. Sucov HM, Dyson E, Gumeringer CL. et al. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994;8:1007-18
6. Li J, Chanrion M, Sawey E. et al. Reciprocal Interaction of Wnt and RXR-α Pathways in Hepatocyte Development and Hepatocellular Carcinoma. PLoS One. 2015;10:e0118480
7. Shulman AI, Mangelsdorf DJ. Retinoid X Receptor Heterodimers in the Metabolic Syndrome. N Engl J Med. 2005;353:604-15
8. Nam KN, Mounier A, Fitz NF. et al. RXR controlled regulatory networks identified in mouse brain counteract deleterious effects of Aβ oligomers. Sci Rep. 2016 [Epub ahead of print]
9. McFarland K, Spalding TA, Hubbard D. et al. Low dose bexarotene treatment rescues dopamine neurons and restores behavioral function in models of Parkinson's disease. ACS Chem Neurosci. 2013;4:1430-8
10. Kim MS, Lim do Y, Kim JE. et al. Src is a novel potential off-target of RXR agonists, 9-cis-UAB30 and Targretin, in human breast cancer cells. Mol Carcinog. 2015;54:1596-604
11. Tsai DE, Luger SM, Andreadis C. et al. A phase I study of bexarotene, a retinoic X receptor agonist, in non-M3 acute myeloid leukemia. Clin Cancer Res. 2008;14:5619-25
12. Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269-304
13. Kizaki M, Dawson MI, Heyman R. et al. Effects of novel retinoid X receptor-selective ligands on myeloid leukemia differentiation and proliferation in vitro. Blood. 1996;87:1977-84
14. Altucci L, Rossin A, Hirsch O. et al. Rexinoid triggered differentiation and tumor-selective apoptosis of acute myeloid leukemia by protein kinase A-mediated desubordination of retinoid X receptor. Cancer Res. 2005;65:8754-65
15. Qiu JJ, Zeisig BB, Li S. et al. Critical role of retinoid/rexinoid signaling in mediating transformation and therapeutic response of NUP98-RARG leukemia. Leukemia. 2015;29:1153-62
16. Love JD, Gooch JT, Benko S. et al. The structural basis for the specificity of retinoid-X receptor-selective agonists: new insights into the role of helix H12. J Biol Chem. 2002;277:11385-91
17. Li Y, Zhang Y, Hill J. et al. The Rexinoid LG100268 prevents the development of preinvasive and invasive estrogen receptor negative tumors in MMTV-erbB2 mice. Clin Cancer Res. 2007;13:6224-31
18. Cao M, Royce DB, Risingsong R. et al. The rexinoids LG100268 and LG101506 inhibit inflammation and suppress lung carcinogenesis in A/J mice. Cancer Prev Res (Phila). 2016;9:105-14
19. Gómez E, Rodríguez A, Muñoz M. et al. Development and quality of bovine morulae cultured in serum-free medium with specific retinoid receptor agonists. Reprod Fertil Dev. 2008;20:884-91
20. Rendi MH, Suh N, Lamph WW. et al. The selective estrogen receptor modulator arzoxifene and the rexinoid LG100268 cooperate to promote transforming growth factor beta-dependent apoptosis in breast cancer. Cancer Res. 2004;64:3566-71
21. Lo-Coco F, Avvisati G, Vignetti M. et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111-21
22. Lo-Coco F, Cicconi L, Breccia M. Current standard treatment of adult acute promyelocytic Leukaemia. Br J Haematol. 2016;172:841-54
23. Breccia M, Minotti C, Latagliata R. et al. Influence of time to complete remission and duration of all-trans retinoic acid therapy on the relapse risk in patients with acute promyelocytic leukemia receiving AIDA protocols. Leuk Res. 2013;37:383-5
24. Tallman MS, Andersen JW, Schiffer CA. et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337:1021-8
25. Luscher B, Vervoorts J. Regulation of gene transcription by the oncoprotein MYC. Gene. 2012;494:145-60
26. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976-90
27. Prathapam T, Tegen S, Oskarsson T. et al. Activated Src abrogates the Myc requirement for the G0/G1 transition but not for the G1/S transition. Proc Natl Acad Sci USA. 2006;103:2695-700
28. Wang H, Mannava S, Grachtchouk V. et al. c-Myc depletion inhibits proliferation of human tumor cells at various stages of the cell cycle. Oncogene. 2008;27:1905-15
29. Fu X, Tan D, Hou Z. et al. miR-338-3p is down-regulated by hepatitis B virus X and inhibits cell proliferation by targeting the 3'-UTR region of CyclinD1. Int J Mol Sci. 2012;13:8514-39
30. Musgrove EA, Caldon CE, Barraclough J. et al. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558-72
31. Vitale I, Galluzzi L, Castedo M. et al. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385-92
32. Necochea-Campion Rd, Chen CS, Mirshahidi S. et al. Clinico-pathologic relevance of survivin splice variant expression in cancer. Cancer Lett. 2013;339:167-74
33. Carter BZ, Qiu Y, Huang X. et al. Survivin is highly expressed in CD34(+)38(-) leukemic stem/progenitor cells and predicts poor clinical outcomes in AML. Blood. 2012;120:173-80
34. Karami H, Baradaran B, Esfahani A. et al. siRNA-mediated silencing of survivin inhibits proliferation and enhances etoposide chemosensitivity in acute myeloid leukemia cells. Asian Pac J Cancer Prev. 2013;14:7719-24
35. Adida C, Recher C, Raffoux E. et al. Expression and prognostic significance of survivin in de novo acute myeloid leukaemia. Br J Haematol. 2000;111:196-203
36. Zhou J, Bi C, Janakakumara JV. et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009;113:4052-62
37. Veres B, Radnai B, Gallyas F. et al. Regulation of kinase cascades and transcription factors by a poly (ADP-ribose) polymerase-1 inhibitor, 4-hydroxyquinazoline, in lipopolysaccharide-induced inflammation in mice. J Pharmacol Exp Ther. 2004;310:247-55
38. Siendones E, Barbarroja N, Torres LA. et al. Inhibition of Flt3-activating mutations does not prevent constitutive activation of ERK/Akt/STAT pathways in some AML cells: a possible cause for the limited effectiveness of monotherapy with small-molecule inhibitors. Hematol Oncol. 2007;25:30-7
39. Han BS, Wei W, Hua FY. et al. Requirement for ERK activity in sodium selenite-induced apoptosis of acute promyelocytic leukemia-derived NB4 cells. J Biochem Mol Biol. 2007;40:196-204
40. Chan KT, Li K, Liu SL. et al. Cucurbitacin B inhibits STAT3 and the Raf/MEK/ERK pathway in leukemia cell line K562. Cancer Lett. 2010;289:46-52
41. Barbarroja N, Siendones E, Torres LA. et al. MEK inhibition induces caspases activation, differentiation blockade and PML/RARalpha degradation in acute promyelocytic leukaemia. Br J Haematol. 2008;142:27-35
42. Geest CR, Coffer PJ. MAPK signaling pathways in the regulation of hematopoiesis. J Leukoc Biol. 2009;86:237-50
43. Wang Y, Liu L, Zhou D. Inhibition of p38 MAPK attenuates ionizing radiation-induced hematopoietic cell senescence and residual bone marrow injury. Radiat Res. 2011;176:743-52
44. Xiao Y, Zou P, Wang J. et al. Lower phosphorylation of p38 MAPK blocks the oxidative stress-induced senescence in myeloid leukemic CD34(+)CD38 (-) cells. J Huazhong Univ Sci Technolog Med Sci. 2012;32:328-33
45. Sanchez PV, Glantz ST, Scotland S. et al. Induced differentiation of acute myeloid leukemia cells by activation of retinoid X and liver X receptors. Leukemia. 2014;28:749-60
Author contact
![]() Corresponding author: BeiZhong Liu, Department of Laboratory Medicine, Chongqing Medical University, 1#, Yixueyuan Road, Chongqing, 400016, China. Tel: +86 18716474304, Fax: +86 023-68485006; E-mail: liubeizhongedu.cn
Corresponding author: BeiZhong Liu, Department of Laboratory Medicine, Chongqing Medical University, 1#, Yixueyuan Road, Chongqing, 400016, China. Tel: +86 18716474304, Fax: +86 023-68485006; E-mail: liubeizhongedu.cn