Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(5):365-373. doi:10.7150/ijms.14989 This issue Cite
Research Paper
EphB4/ephrinB2 Contributes to Imatinib Resistance in Chronic Myeloid Leukemia Involved in Cytoskeletal Proteins
Lin Li1,*, Na Xu1,*, Jin-fang Zhang2, Lu-lu Xu1, Xuan Zhou1, Bin-tao Huang3, Yu-ling Li1, Xiao-li Liu1, ![]()
1. Department of Hematology, Nanfang Hospital, Southern Medical University, 1838 North Guangzhou Ave, Guangzhou 510515, Guangdong, China
2. Department of Paediatric Hematology and Oncology, Clinical Center of Tumor Therapy, Guangdong General Hospital, Guangdong Academy of Medical Sciences, 510000, China
3. Department of Hematology, The Affiliated Hospital of Inner Mongolia Medical University, 1 Tongdao Avenue North, Hohhot 010059, China
* Lin Li and Na Xu contributed equally to this work and should be considered as co-first authors.
Received 2016-1-16; Accepted 2016-4-12; Published 2016-4-28
Abstract
Introduction: The mechanism of EphB4/ephrinB2 in the resistance of chronic myelogenous leukemia to imatinib keeps unknown.
Methods: The imatinib resistant chronic myelogenous leukemia cell line-K562-R, was established. EphB4 receptor expression was detected in patients and resistant cells. Cell migration and drug sensitivity were tested in the EphB4 knockdown cells and mouse models.
Results: The EphB4 receptor was over-expressed in blast crisis patients compared to chronic phase patients. The level of EphB4 receptor expression was associated with a complete cytogenetic response within 12 months. Enhanced expression of the EphB4 receptor was detected in the K562-R cells. EphB4 knockdown inhibited cell migration ability and restored sensitivity to imatinib in vitro and in vivo. Restored sensitivity to imatinib was observed in K562-R cells, along with increased levels of phospho-EphB4 and decreased phosphorylation levels of RhoA, Rac1, and Cdc42.
Conclusion: Our study illustrates that aberrant activation of EphB4/ephrinB2 may mediate chronic myeloid leukemia resistance involved in cytoskeletal proteins.
Keywords: Chronic myelogenous leukemia, imatinib, EphB4/ephrin B2, shRNA
Introduction
Imatinib (IM) is used as the standard chemotherapy drug for many chronic myelogenous leukemia (CML) patients and can significantly prolong a patient's survival. However, IM drug resistance occurs in 20-30% of patients and results in a progressive outcome. Although second generation kinase inhibitors can overcome this BCR-ABL-dependent resistance, their effects are limited [1]. TKI-resistant CML is more complex than the original spontaneous IM resistance. BCR-ABL-dependence also contributes to resistance [2].
Signal transduction through the erythropoietin producing hepatoma (Eph) amplified sequence receptors binding to their cell-surface ephrin ligands and have been implicated in hematopoiesis and the growth of various cancer cells [3].The binding of an ephrin ligand to its respective Eph receptor activates a bi-directional signal (as well as cell adhesion), thereby affecting cell proliferation and cell-fate determination in a number of key biological processes, including angiogenesis and hematopoiesis [4-6]. The Eph receptors (which are phosphorylated on two conserved tyrosines) enable efficient kinase activation in the juxta-membrane domain, which modulates many adaptors and effectors including non-receptor tyrosine kinases of the Src and Abl families (particularly Rho and Ras family GTPases). This activation may also regulate the molecular expression of other receptor tyrosine kinase (RTK)family members [5, 7, 8]. These crucial regulators assist most RTK families in controlling cell growth and migration, which ph receptors use to inhibit cell proliferation and survival.
Some studies have indicated that ephrinB2-Fc activated EphB4 receptors could decrease cell viability and suppress cell invasion in breast and colon cancer [9-11]. Previous reports have also found that increased EphB4 expression levels are related to poor prognoses in breast cancer [12]. Recent findings have shown that EphB4/ephrinB2 signaling may be involved in the pathology of leukemia [3], and over expression of EphB4 has been associated with drug resistance in Ph+ acute lymphocytic leukemia (ALL) [13]. It also has been shown that EphB4 promotes cell adhesion-mediated drug resistance by interacting with the extracellular matrix and modulating Rho family members.
Our previous study has established the EphB4 knockdown and IM resistant CML cell line (K562-R-EphB4-sh) and found that homoharringtonine overcomes imatinib resistance by blocking the EphB4/RhoA pathway in K562-R [14]. To determine the role and mechanism of EphB4/ephrinB2 in IM resistance to CML, we investigated the role of EphB4 in IM resistant CML, and we further explored the related molecules and the down-stream signaling pathway of EphB4/ephrinB2.
Materials and Methods
Study Population
Clinical samples were taken from the bone marrow of chronic myelogenous leukemia chronic phase (CML-CP) patients (n=22), as well as chronic myelogenous leukemia blast crisis (CML-BC) patients (n=6) to produce bone marrow (BM) cells in the first diagnosis. All of the patients had received IM therapy, and CML-BC patients were resistant to imatinib. The clinical diagnosis of CML was evaluated by the National Comprehensive Cancer Network. All patients agreed to provide their written informed consent for the use of their samples. The protocol was approved by the Ethics Committee of the Nanfang Hospital, Southern Medical University.
Cell Culture
K562 wild type cells (American Type Culture Collection, Manassas, VA, USA) were cultured in a RPMI-1640 medium supplemented with 10% Fetal Bovine Serum (FBS)(Sigma-Aldrich, St. Louis, MO, USA) at 37˚C, 5% CO2. K562 wild type cells were incubated with IM (initial at 0.05 µM) to establish IM resistance using the step-wise method described previously [14]. IM (Sigma, USA) concentration was increased by 0.05 µM every ten or twenty days until reaching 2.8 µM. The surviving cells were then cultured in RPMI-1640/10% FBS at 37˚C, 5% CO2 in the absence of IM for approximately three days. Cells were re-examined for sensitivity to IM and resistant K562 cells were labeled as K562-R. K562-R-EphB4-sh represented the established IM resistant cells with EphB4 knockdown by shRNA from our previous study [14].
Real Time PCR
Total RNA of patient samples and cell lines was extracted with the Trizol reagent (Invitrogen, Carlsbad, CA, USA) and stored at -80˚C. All RT-PCR primers were designed with Primer 5.0 and listed below:
EphB4 forward primer: 5'-ACCTCCATTCCTGCGGCTAA-3'; Reverse primer: 5'-AGACGAGGTTGCTGTTGACT-3'.
GAPDH forward primer: 5'-GAGGGGTGATGTGGGGAGTA-3'; Reverse primer: 5'-GAGCTTCCCGTTCAGCTCAG-3'.
PCR conditions were: initial 95˚C, 15 min; denaturation 94˚C, 15 s; annealing 60˚C 30 s and extension 72˚C, 30 s. The number of cycles was 35-45.
Western blot analysis
Proteins were firstly extracted using the RIPA lysis buffer with adding halt protease and the phosphatase inhibitor (Thermo Fisher Scientific, Boston, MA, USA). The concentrations of proteins were measured with the bicinchoninic acid (BCA) method. Proteins were then separated by SDS-PAGE and transferred onto nitrocellulose membranes following 2 hours blocking with 5% bovine serum albumin (BSA). The membrane was incubated with primary antibody at 4˚C overnight and then continually incubated with HRP-conjugated goat anti-rabbit (1:2000) or goat anti-mouse (1:5000) (Cell signaling, Berverly, MA, USA) immunoglobulin diluted in 5% BSA for l hour. Blots were washed again, followed by an enhanced chemiluminescence assay (Cell signaling) to visualize the secondary antibody.
Apoptosis assessment and cell cycle assay
K562, K562-R-EphB4-sh, and K562-R cells were collected, washed twice, and incubated with PE Annexin V in a buffer containing 7-AAD for 15 min at room temperature (BD Pharminigen, San Diego, CA, USA.). Cells were then examined by flow cytometry. Cells (5×105 cells/ml) were seeded in 24-well plates and cultured in l ml media for 24 hours. The exponentially growing cells were then incubated with IM (0.5 µM). Cells were pelleted and then fixed for 1 h at 4˚C by adding 500 µL of fixation solution. The fixed cells were pelleted and analyzed by flow cytometry (FACS)(BD Pharminigen).
Cell migration assay
Cell migration was detected with commercial transwell assay (Corning, Tewksbury, MA, USA). Briefly, 10% (v/v) fetal calf serum 1640-mediumwas added to the lower chamber, and 3×104cells were suspended in serum-free media and added to the upper chamber following 4 h incubation supplied with 5% CO2 at room temperature. Then, the upper chamber was detached from the assay, and the cells were collected from the bottom chamber. Total number of cells was counted at three randomly selected fields.
Cell viability assay
Cell viability of K562, K562-R-phB4-sh, and K562-R cells were measured using a cell counting kit-8 (CCK-8) (Sigma). 5×104 cells/well were seeding into 96-well micro-plate containing in 100 µl media and followed a series of concentrations of IM (0-8 µM) treatment. After 24 hours of culture, 10 µl CCK-8 solution was added into each well and incubated for 2 hours at room temperature. Cell viability was detected using a microplate reader (Bio-rad 680, Hercules, CA, USA) at 450nm optical density (OD) and quantified as [(OD treated -OD blank)/(ODcontrol-OD blank]× 100%. The IC50 value of each cell line was calculated from the cell inhibition rate.
Xenograft models
K562 parental cells, K562-R-EphB4-sh, and K562-R cells (1x107/ml) were subcutaneously injected into BALB/C nude mice, (female nude mice 4-5 weeks of age; SLRC laboratory animal center, China) to build xenograft models. On day 30 after implantation, tumors reached an average size of over 1000 mm. As determined by histopathology, green fluorescent protein (GFP) positive cells in the xenograft tumor were found in the K562-R-EphB4-sh cell group. Then, all nude mice received an oral standard-dose of IM (91 µg/g daily for 30 days) treatment. All animal experiments were conducted according to the policy and procedures provided by the Committee for Animal Research and Ethics and were approved by the Ethics Committee of Nanfang Hospital, Southern Medical University.
Enzyme-linked immunosorbent assay (ELISA)
The phosphorylation level of EphB4 in each cell line was detected by phosphorylation ELISA (Abcam, Cambridge, UK). The anti-human EphB4 antibody specifically recognizes and binds human EphB4 proteins (including the phosphorylated and non-phosphorylated proteins) and was immobilized on 96-well micro titer plates as the capture antibody. Cell lysate was then added to the plates. Plates were washed to remove unbound proteins, and HRP-conjugated anti-tyrosine phosphorylated antibodies specific for phosphorylated proteins were added. After the plates were washed again, chromogenic substrate was added and the absorbance of the substrate was measured at 450 nm.
Statistical analysis
Data were presented with the mean ± standard deviation (SD) from three independent experiments. SPSS13.0 statistical software (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. Paired t test or LSD method were used for the comparison between groups and within groups. Pearson correlation coefficients were performed to assess the correlation between EphB4 mRNA and BCR-ABL transcript levels. P < 0.05 was considered to be statistically significant.
Results
Association of EphB4 expression with clinical stage and IM drug response in CML patients
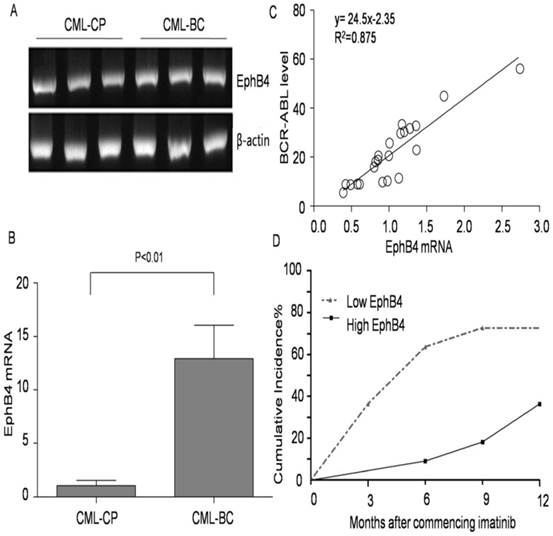
The expression levels of EphB4 from CML-CP and CML-BC patients are shown in Fig. 1A. Our results also show that EphB4 mRNA levels in CML patients significantly increased according to clinical stages (P<0.01) (Fig. 1B). We evaluated the imatinib response in 22 CML-CP patients (400 mg/d or 600 mg/d). EphB4 mRNA levels in cancer cells correlated with the corresponding BCR-ABL transcript levels at 3 months in each case (y=24.5x-2.35 R2=0.875, P<0.01), as shown in Fig. 1C. Most of the IM treated patients, 63.6% (14/22), achieved complete cytogenetic response (CCyR) in 12 months (Fig. 1D). These patients were then equally divided into two groups according to the relative EphB4 mRNA median level (0.988). The 12-month CCyR rate in the low EphB4 group was 72.7%, and the CCyR rate was 36.3% in the high EphB4 group (P<0.01).
Association of EphB4 expression with clinical stage in CML patients. A. Expression of EphB4 in the bone marrow samples of CML patients. B. Comparison of EphB4 mRNA in CML-CP and CML-BC patients. C. Correlation between EphB4 mRNA levels in cancer cells with BCR-ABL mRNA levels in CML-CP patients. D. Patients' 12-month CCyR rate was analyzed after commencing imatinib treatment.
Regulation of EphB4 on cell migration and viability
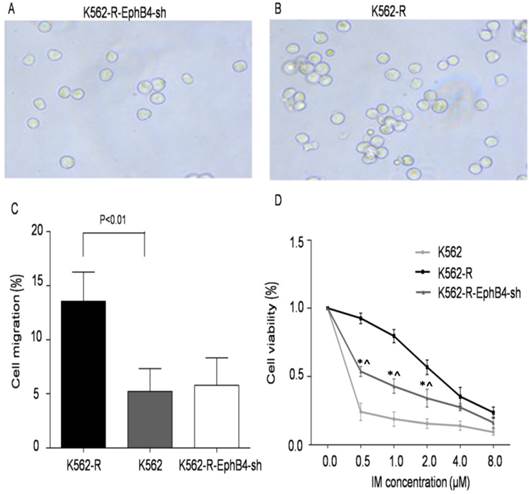
Previous publications have shown that EphB4 regulates the activity of the actin cytoskeleton. Thus, we tested whether EphB4 expression levels influenced K562 cell migration. The results demonstrate that cell migration in K562-R-EphB4-sh was significantly decreased (57.4±3.01) compared with K562-R, but it was not significantly statistically different from K562 (Fig. 2A, 2B and 2C). This indicates that EphB4 knockdown impaired the migration of K562-R cells in vitro. Furthermore, we compared K562, K562-R, and K562-R-EphB4-sh in terms of cell viability, apoptosis rate, and cell cycle. The cell viability of K562-R-EphB4-sh cells was less than K562-R, but it was stronger than K562 after treatment with different concentrations of the IM resistant medium (Fig. 2D).
Regulation of EphB4 on cell migration and viability. A and B. Effects of knockdown of EphB4 on the migration of K562-R cells. Representative images of the migrated cells in the transwell assays. C. Comparison of cell migration rate to the control values of K562-R cells, measured with in a transwell assay. D. Comparison of cell viability of K562/K562-R/K562-R-EphB4-sh in different IM concentrations by the CCK8 assay (* P<0.01, compared to K562; ^ P<0.01, compared to K562-R).
Regulation of EphB4 on cell apoptosis and cell cycle distribution
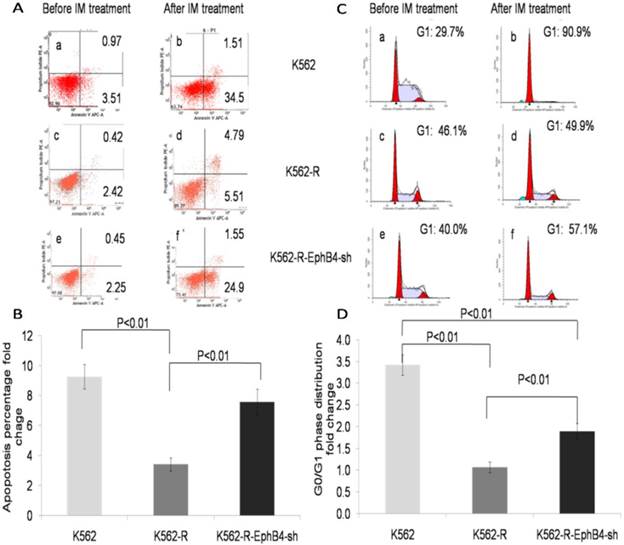
The apoptosis rate of K562-R-EphB4-sh cells was much higher than that of K562-R, but it was lower than that of K562 (Fig. 3A and 3B) after treatment with IM (1µM) for 36 hours. In addition, IM induced increase of cells in the G0/G1 phase and no such effect was observed in K562R cells, while The G0/G1 cell arrest was reappeared in K562-R-EphB4-sh cells following IM treatment. (Fig. 3C and 3D).
Regulation of EphB4 on cell apoptosis and cell cycle distribution. A. Apoptosis distribution of experimental cells (a-b: K562 cell line; c-d: K562-R cell line; e-f: K562-EphB4-sh cell line; a, c, e: before IM treatment; b, d, f: after IM treatment). B. Apoptosis percentage analysis of the three cell lines. C. Cell cycle analysis of the three cell lines (a-b: K562 cell line; c-d: K562-R cell line; e-f: K562-EphB4-sh cell line; a, c, e: before IM treatment; b, d, f: after IM treatment); D. G0/G1 phase distribution analysis of the three cell lines.
Knockdown of EphB4 prevents leukemic cell resistance to IM in vivo
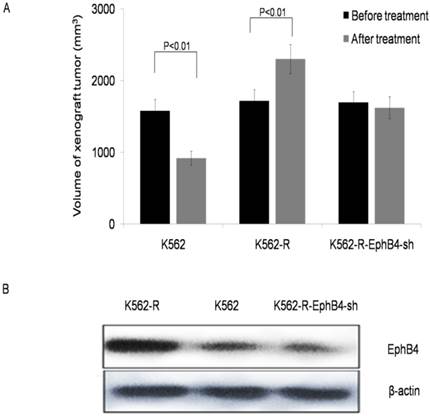
To evaluate the effect of EphB4 on CML resistance in vivo, we created xenograft tumors in BALB/C nude mice. Fig.4A shows the changes in tumor volume of three groups of xenograft tumors before and after treatment with IM. All nude mice in the K562-R xenograft died; 2 and 4 nude mice died in the K562 and K562-R-EphB4-sh xenograft groups, respectively, during the period of treatment (30 days). Analysis of xenograft tissue demonstrated that the highest levels of EphB4 receptor expression were found in K562-R xenograft tissue, while the lowest was found in K562-R-EphB4-sh xenograft tissue (Fig. 4B).
Knockdown of EphB4 prevents leukemic cell resistance to IM in vivo. A. Comparison of volume in three groups of xenograft tumors before and after treated IM. B. Comparison of the expression of EphB4 proteins in xenograft tissue.
Ephrin B2-Fc restored K562-R cells sensitivity to IM
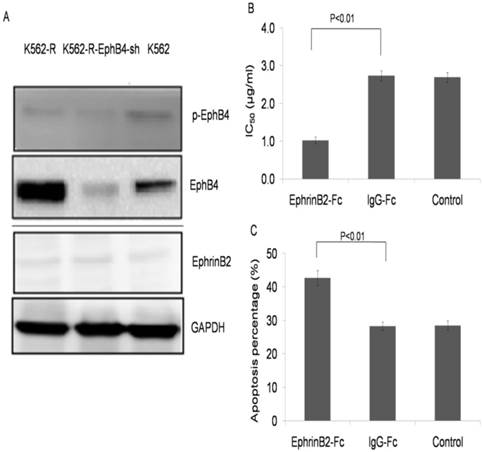
Low expression of phospho-EphB4 and the ephrinB2 ligand were observed in each cell line (Fig. 5A). K562-R cells were treated with ephrinB2-Fc (4µg/ml), IgG-Fc (0.4µg/ml) or the control for 30 min. Then, the cells were exposed to IM at different concentrations (0, 0.625, 1.25, 2.5, 5.0, 10 and 20µM) for 24 hours. CCK-8 results showed an IC50 of 1.07±0.0391 µg/ml for the ephrinB2-Fc group, 2.69±0.145 µg/ml for the IgG-Fc group, and 2.66±0.102 µg/ml for the blank control (Fig. 5B). The apoptosis rate was 42.7%±0.951 for the ephrinB2-Fc group treated with IM (2.7µM, 24 hours).This was higher when compared with the IgG-Fc group (28.8% ± 1.64) or the control group (28.9% ±1.49) (P<0.01) (Fig. 5C and Supplementary Fig.1).
EphrinB2-Fc restored K562-R cells sensitivity to IM. A. Expression of EphB4 and p-EphB4 in K562, K562-R and K562-R-EphB4-sh cell lines. Expression of ephrinB2 in K562, K562-R and K562-R-EphB4-sh. Expression of proteins analyzed by Western blotting. B. Comparison of IC50 in K562-R, ephrinB2-Fc (4µg/ml), K562-R+ IgG-Fc (0.4µg/ml), and control groups in different concentrations of IM by CCK8 assay. C. Apoptosis percentage for the K562-R cell line treatment with EphrinB2-Fc, IgG-Fc, and blank control by flow cytometry.
EphrinB2 enhanced EphB4 was phosphorylated with decreased expression of EphB4 protein in a dose dependent manner
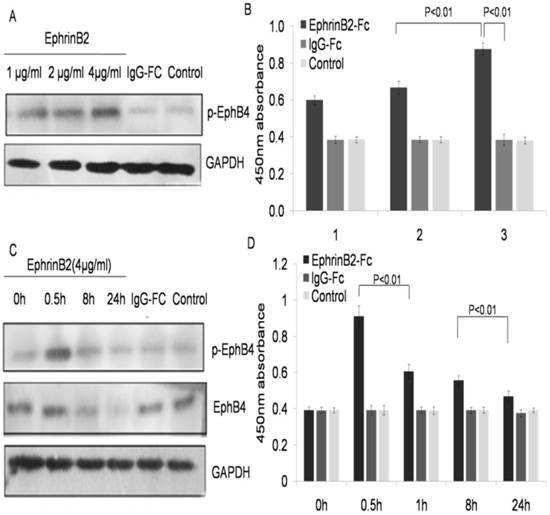
The EphB4 receptor changes were identified by stimulating the ephrinB2 ligand. K562-R cells treated with EphrinB2-Fc (l, 2 and 4µg/ml), IgG-Fc (0.4µg/ml), or the blank control for 30 min. The level of phosphorylation for EphB4 was examined in each treated group by ELISA. The results showed that the phosphorylation of EphB4 increased in the ephrinB2-Fc group compared with both the IgG-Fc group and the blank control in a dose-dependent manner (Fig. 6A and 6B). Furthermore, we studied the change of EphB4 at different time points after stimulation by the ephrin-B2 ligand. The results showed that the phosphorylation level of EphB4 in K562-Rhad significantly increased after treatment with ephrinB2-Fc (4µg/ml) for 0.5h, but it began to decrease after l hour and reached its minimum after 24 hours. However, the expression level of the EphB4 protein did not significantly increase in K562-R cells treated with ephrinB2-Fc (4µg/ml). Rather, it began to decrease after 1 hour and reached its minimum after 24 hours (P<0.01) (Fig. 6C and 6D). There were no significant changes in the two control groups at each time point.
Ephrin B2 enhanced EphB4 phosphorylation and decreased expression of EphB4 protein in a dose dependent manner. A. Phosphorylation of EphB4 in the K562-R cell line after treatment with different concentrations of soluble ephrinB2 protein by western blot. B. Comparison of 450nm absorbance in K562-R cells after treatment with different concentrations of soluble EphrinB2 protein. 1-3 groups represent: 1. EphrinB2-Fc (1 µg/ml), IgG-Fc (0.1 µg/ml); 2. EphrinB2-Fc (2µg/ml), IgG-Fc (0.2µg/ml); 3. EphrinB2-Fc (4µg/ml), IgG-Fc (0.4µg/ml). The blank control had no treatment for each group. C. Expression and phosphorylation of EphB4 in the K562-R cell line following treatment with the soluble ephrinB2 (4µg/ml) protein at a different time by western blot. D. Comparison of 450nm absorbance in the K562-R cell line treated with the soluble ephrinB2 (4µg/ml) protein at a different time.
Phosphorylation of small GTPases in vitro and in vivo
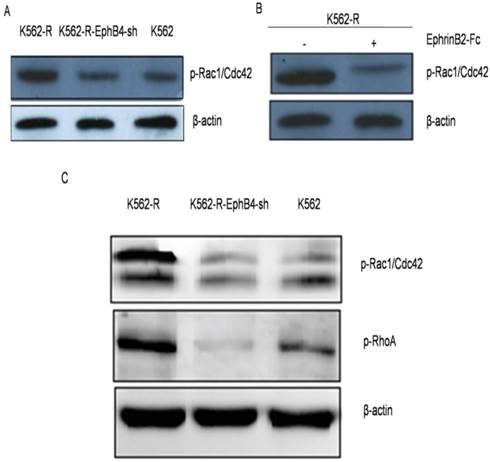
We further examined the expression of EphB4 related proteins in vitro. The expression of phospho-RhoA/Racl/Cdc42 significantly decreased after knockdown of EphB4 in K562-R cells (Fig. 7A). After treatment with ephrinB2-Fc, the expression of phospho-RhoA/Racl/Cdc42 proteins in K562-R cells significantly decreased (Fig. 7B). In vivo, phosphorylation of Racl+Cdc42 and RhoA had the highest expression in the K562-R xenograft tumor (Fig. 7C).
Phosphorylation of small GTPases in vitro and in vivo. A. Phosphorylation level of Rho-GTPases (Rho A, Rac and Cdc42) in three cell lines by western blot. B. Expression of phospho-RhoA/Racl/Cdc42 proteins in K562-R after treatment with EphrinB2-Fc. C. Phosphorylation level of Rho-GTPases (RhoA, Rac and Cdc42) in xenograft tumors.
Discussion
EphB4/ephrins play important roles in oncogenesis and in tumor growth progression [15,16]. Many studies have indicated that they can work as either a prognostic factor or potential treatment target [17-19]. Tomasz and his team confirmed that patients with shorter RFS had decreased EphB4 expression in non-M3 AML patients [20]. Suzuki M reported that the overexpression of EphB4 was related to drug resistance in Ph+ acute lymphocytic leukemia [13]. Our study investigated the expression of Eph B4 mRNA in CML patients and identified a dramatically higher expression of EphB4 receptors in CML-BP patients compared with CML-CP patients. The amounts of EphB4 receptor expression were also strongly associated with an adverse cytogenetic response.
Our results demonstrate that EphB4 was over expressed in K562-R cells. Further studies showed that ephrinB2-Fc stimulation restored cell sensitivity to IM in EphB4 knockdown cells. The same effect was observed in K562-R-EphB4-sh xenograft nude mice with oral IM treatment. Together these results indicate that the EphB4 pathway is partially responsible for IM resistance. However, how the EphB4 receptor contributes to IM resistance in CML remains elusive. Several studies have demonstrated the ability of Eph receptors to activate different Rho-GTPases, such as RhoA and Rac1/Cdc42 [21]. The Rho family plays an essential role in IM resistant CML through BCR-ABL non-kinase pathways [22]. These studies suggest that EphB4 may contribute to IM resistance in CML at least partly through Rho signaling and Rho-mediated actin cytoskeleton regulation, and knockdown of EphB4 contributes to partial recovery of IM sensitivity.
In this study, our results showed that knocking down EphB4 expression resulted in resistant cells that were arrested in the G0 phase following IM treatment. Additionally, knockdown of EphB4 or ephrinB2-Fc significantly stimulated a reduction in RhoA GTPase expression in vitro and in vivo. These results illustrate an association between EphB4 and Rho GTPase. Previous studies had confirmed that the Rho-GTPase family could regulate how Eph receptors could affect the actin cytoskeleton and subsequently disrupt cell shape, adhesion, and migration [5]. In addition, Wang and his colleagues reported that Rac1-GTPase plays an important role in leukemia cell chemotherapy resistance, quiescence maintenance, and in the interaction with the bone marrow microenvironment [1]. Our previous study also demonstrated that HHT contributes to IM sensitivity by blocking the EphB4/RhoA pathway in CML cell lines [14]. Therefore, in advanced CML, Eph causes gradual leukemia cell chemotherapy resistance by up-regulating Rho-GTPase while promoting proliferation and inhibiting apoptosis by up-regulating Ras-GTPases and Akt/mTORC1.
In summary, our study demonstrated that knocking down EphB4 could restore CML cell sensitivity to IM, increase the percentage of drug-induced apoptosis, and decrease invasive ability through a GTPase (RhoA/Racl/Cdc42)-dependent mechanism. EphrinB2 played a crucial role in tumor inhibition by activating EphB4 downstream signaling. Our studies have thus provided new biomarkers for the development of novel drugs for the disease.
Supplementary Material
Supplementary figure 1.
Acknowledgements
We would like to thank all the authors participating sites. This work was supported by National Natural Science Foundation of China (No.81300435 and No.81170521) and the Science and Technology Planning Project of Guangdong Province, China (2014A020211019).
Competing interest
No conflict of interest exists in the submission of this manuscript, and it is approved by all of the authors for publication.
References
1. Kujawski L, Talpaz M. Strategies for overcoming imatinib resistance in chronic myeloid leukemia. Leuk Lymphoma. 2007;48:2310-22
2. Jabbour EJ, Cortes JE, Kantarjian HM. Resistance to tyrosine kinase inhibition therapy for chronic myelogenous leukemia: A clinical perspective and emerging treatment options. Clin Lymphoma Myeloma Leuk. 2013;13:515-29
3. Takahashi Y, Itoh M, Nara N. et al. Effect of EPH-ephrin signaling on the growth of human leukemia cells. Anticancer Res. 2014;34:2913-8
4. Pasquale EB. Eph receptor signaling casts a wide net on cell behavior. Nature Rev Mol Cell Biol. 2005;6:462-75
5. Pasquale EB. Eph receptors and ephrins in cancer: Bidirectional signaling and beyond. Nat Rev Cancer. 2010;10:165-80
6. Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol. 2013;5:a009159
7. Choura M, Rebai A. Receptor tyrosine kinases: from biology to pathology. J Recept Signal Transduct Res. 2011;31:387-94
8. Jorgensen C, Sherman A, Chen GI. et al. Cell-specific information processing in segregating populations of Eph receptor ephrin-expressing cells. Science. 2009;326:1502-9
9. Noren NK, Pasquale EB. Paradoxes of the EphB4 receptor in cancer. Cancer Res. 2007;67:3994-7
10. Dopeso H, Mateo-Lozano S, Mazzolini R. et al. The receptor tyrosine kinase EPHB4 has tumor suppressor activities in intestinal tumorigenesis. Cancer Res. 2009;69:7430-8
11. Ronsch K, Jager M, Schöpflin A. et al. Class I and III HDACs and loss of active chromatin features contribute to epigenetic silencing of CDX1 and EPHB tumor suppressor genes in colorectal cancer. Epigenetics. 2011;6:610-2
12. Brantley-Sieders DM, Zhuang G, Hicks D. et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Invest. 2008;118:64-78
13. Suzuki M, Abe A, Imagama S. et al. BCR-ABL-Independent and RAS/MAPK pathway dependent form of imatinib resistance in Ph-positive acute lymphoblastic leukemia cell line with activation of EphB4. Eur J Haematol. 2010;84:229-38
14. Huang BT, Zeng QC, Zhao WH. et al. Homoharringtonine contributes to imatinib sensitivity by blocking the EphB4/RhoA pathway in chronic myeloid leukemia cell lines. Med Oncol. 2014;31:836
15. Mosch B, Reissenweber B, Neuber C. et al. Eph receptors and ephrin ligands: Important players in angiogenesis and tumor angiogenesis. J Oncol. 2010;2010:135285
16. Boyd AW, Bartlett PF, Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat Rev Drug Discov. 2014;13:39-62
17. Li D, Liu S, Liu R. et al. Targeting the EphB4 receptor for cancer diagnosis and therapy monitoring. Mol Pharm. 2013;10:329-36
18. Day BW, Stringer BW, Boyd AW. Eph receptors as therapeutic targets in glioblastoma. Br J Cancer. 2014;111:1255-61
19. Edwards CM, Mundy GR. Eph receptors and Ephrin signaling pathways: a role in bone homeostasis. Int J Med Sci. 2008;5:263-72
20. Wrobel T, Pogrzeba J, Stefanko E. et al. Expression of Eph A4, Eph B2, and Eph B4 receptors in AML. Pathol Oncol Res. 2014;20:901-7
21. Yang NY, Pasquale EB, Owen LB. et al. The EphB4 Receptor-tyrosine Kinase promotes the migration of melanoma cells through Rho-mediated actin cytoskeleton reorganization. J Biol Chem. 2006;281:32574-86
22. Kuzelová K, Hrkal Z. Rho-signaling pathways in chronic myelogenous leukemia. Cardiovasc Hematol Disord Drug Targets. 2008;8:261-7
Author contact
![]() Corresponding author: Xiao-li Liu, Department of Hematology, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, China. Phone: +86 (020) 6164-1616; Fax: +86 (020) 6164-1616; E-mail: lxl2405com
Corresponding author: Xiao-li Liu, Department of Hematology, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, China. Phone: +86 (020) 6164-1616; Fax: +86 (020) 6164-1616; E-mail: lxl2405com