Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(5):357-364. doi:10.7150/ijms.14592 This issue Cite
Research Paper
Autophagy Has a Beneficial Role in Relieving Cigarette Smoke-Induced Apoptotic Death in Human Gingival Fibroblasts
Moon-Soo Kim1, Jeong-Won Yun1, Jin-Ho Park1, Bong-Wook Park1, Young-Hoon Kang1, Young-Sool Hah2, Sun-Chul Hwang3, Dong Kyun Woo4, ![]() , June-Ho Byun1,
, June-Ho Byun1, ![]()
1. Department of Oral and Maxillofacial Surgery, Gyeongsang National University School of Medicine and Gyeongsang National University Hospital, Institute of Health Sciences, Gyeongsang National University, Chilam-dong, Jinju, 660-702, Republic of Korea
2. Clinical Research Institutue of Gyeongsang National University Hospital, Jinju, Republic of Korea
3. Department of Orthopaedic Surgery, Institute of Health Sciences, Gyeongsang National University School of Medicine, Jinju, Republic of Korea
4. College of Pharmacy and Research Institute of Pharmaceutical Sciences, Gyeongsang National University, Jinju, Republic of Korea
Received 2015-12-3; Accepted 2016-4-12; Published 2016-4-27
Abstract
The deleterious role of cigarette smoke has long been documented in various human diseases including periodontal complications. In this report, we examined this adverse effect of cigarette smoke on human gingival fibroblasts (HGFs) which are critical not only in maintaining gingival tissue architecture but also in mediating immune responses. As well documented in other cell types, we also observed that cigarette smoke promoted cellular reactive oxygen species in HGFs. And we found that this cigarette smoke-induced oxidative stress reduced HGF viability through inducing apoptosis. Our results indicated that an increased Bax/Bcl-xL ratio and resulting caspase activation underlie the apoptotic death in HGFs exposed to cigarette smoke. Furthermore, we detected that cigarette smoke also triggered autophagy, an integrated cellular stress response. Interesting, a pharmacological suppression of the cigarette smoke-induced autophagy led to a further reduction in HGF viability while a pharmacological promotion of autophagy increased the viability of HGFs with cigarette smoke exposures. These findings suggest a protective role for autophagy in HGFs stressed with cigarette smoke, highlighting that modulation of autophagy can be a novel therapeutic target in periodontal complications with cigarette smoke.
Keywords: cigarette smoke, oxidative stress, apoptosis, autophagy, gingival fibroblast
Introduction
Cigarette smoke has been implicated as an important risk factor in various diseases including periodontal complications [1,2]. These pathogenic effects of cigarette smoke on diverse human tissues and cells are mainly from the multiple toxic compounds present in cigarette smoke. To date, cigarette smoke is known for containing more than 6,000 compounds, of which more than 150 are reported as toxic chemicals that contribute to the pathogenesis of many human diseases [3]. Although the mechanisms by which cigarette smoke causes diseases remain to be fully determined, recent studies suggest that toxic compounds in cigarette smoke not only initiate and exacerbate tissue injuries but may also impair reparative processes, for example, inflammatory responses [4].
Apoptosis, or programmed cell death, is a tightly regulated process consisting of complex biochemical cascades that involve the caspase activation through either extrinsic (death receptor), intrinsic (mitochondrial) or perforin/granzyme pathways [5,6]. Apoptosis occurs normally during development and aging as a homeostatic mechanism to maintain cell populations in tissues. In addition, apoptosis occurs as a defense mechanism such as in immune reactions or when cells are damaged by noxious agents [5]. Thus, an inappropriate apoptosis has been implicated in many pathologic conditions, such as neurodegenerative disorder and cancer [7,8]. Furthermore, recent studies have reported that cigarette smoke can induce apoptosis in several cell types [9-12]. Most studies showed that cigarette smoke inhibits cell proliferation through these apoptotic pathways, whereas there have been some reports that are contrary [8,13,14].
Autophagy, a regulated catabolic process, occurs not only continuously at basal levels for homeostatic turnover of cytoplasmic components but is also induced in response to a variety of stress stimuli including oxidative stress [12,15]. During autophagy, the cellular components are sequestered in the vesicular system and then delivered to lysosomes for degradation and recycling of biogenic components. Thus, autophagy can be viewed as a cellular attempt to survive through the removal of damaged organelles. However, intriguingly, the induction of autophagy could also be part of the cellular program leading to cell death [16,17]. Thus, failures of the cell to regulate autophagy have also been implicated in pathogenesis of cancer, cardiovascular failure, immune disease, skeletal muscle atrophy and neurodegenerative disorders [16,18].
In dental clinics, smoking is one of the most common risk factors regarding post-extraction complications and periodontal diseases. Interestingly, exposure to smoke-derived toxic compounds has been shown to lead to immune dysfunction [19,20]. Regarding dental health, gingival fibroblasts are one of major players in the host immune defense against several pathogens, and play a crucial role in maintaining tissue structure and function [21,22]. Nevertheless, currently, there is limited evidence regarding the effects of cigarette smoke on human gingival fibroblasts. Thus, this study investigated possible effects of cigarette smoke on human gingival fibroblast viability by examining oxidative stress, apoptosis, and autophagy in human gingival fibroblast cultures exposed to cigarette smoke.
Materials and Methods
Reagents and preparation of cigarette smoke extract (CSE)
Dichlo rodihydrofluorescein diacetate (DCF-DA) was purchased from Invitrogen (USA). N-acetyl cysteine (NAC), 3-methyladenine, and rapamycin were purchased from Sigma (USA). Filter cigarettes, each containing 0.1 mg nicotine and 1 mg tar, were used to prepare the cigarette smoke extract (CSE) used in this study. CSE was prepared in a fume hood by bubbling the smoke from a cigarette in a holder attached by rubber tubing to a peristaltic pump. The pump outflow track was attached by rubber tubing to a glass straw submerged in phosphate-buffered saline. Smoke from ten cigarettes was passed through 20 ml PBS to obtain 100% CSE. Extracts were separated into aliquots and stored at -20°C. Final concentrations of 0.5%, 1%, and 2% CSE were used for all experiments in this study. Control extracts were similarly prepared from unlit cigarettes.
Cell cultures and cell viability assay
Normal human gingival fibroblasts (HGFs) obtained from American Type Culture Company ((HGF-1, CRL-2014). HGFs were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, USA) supplemented with 10% fetal bovine serum and 5% penicillin/streptomycin at 37°C in 5% CO2 humidified incubators. Proliferation of HGFs was measured using the Cell Counting Kit (CCK)-8 (Dojindo, Korea). Briefly, 3 × 105 cells were seeded onto a 96-well plate and cultured for 24 or 48 hours in DMEM containing 0% to 2% CSE. After CSE exposures, the cells were treated with 10 µl CCK-8 solution per well, and incubated for 1 hr at 37°C. Formazan dye generation by cellular dehydrogenase activity was then determined by measuring absorbance at 450 nm using a microplate reader (Perkin Elmer, USA). Cell viability was expressed as percent viability of control cells cultured in the absence of CSE.
Preparation of cell lysates and Western blot analysis
Cells were incubated for 30 min in NP-40 lysis buffer [20 mM Tris pH 7.5 containing 140 mM NaCl, 1 mM EDTA, 1% (v/v) Nonidet P-40, 5 μM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), 1.5 nM aprotinin, 10 nM E-64, and 10 nM leupeptin]. The cells were then sonicated and centrifuged at 12,000 × g for 10 min at 4°C to remove insoluble debris. Protein concentration was determined by the Bradford method. Total proteins (30 μg) were resolved on 10% to 12% SDS-PAGE gels. After electrophoresis, proteins were transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA), immunoblotted with primary antibodies [Bax, Bcl-xL, LC3II, and β-actin (Cell Signaling Technology, USA)], and detected with peroxidase-linked antibodies and a Pierce ECL detection system (Thermo Scientific, USA).
Measurement of cellular H2O2 production and apoptosis by flow cytometry
For assessing cellular H2O2 production, HGFs were cultured in DMEM with 10% fetal bovine serum following 0% to 2% CSE treatments and were stained with DCF-DA (Invitrogen, USA) dissolved in DMEM (incubated at 37°C for 20 minutes). Cells then were washed three times with PBS containing 1% fetal bovine serum and resuspended in 200 μL of 1× PBS containing 1% fetal bovine serum. Stained cells (∼10,000 cells per group) then were analyzed by flow cytometry using a FACSCalibur (BD Biosciences, USA). Flow cytometry data were analyzed using a FlowJo software and all experiments were performed in triplicate. For apoptosis assays, HGFs were cultured similarly and double-stained with Annexin V and propidium iodide (PI). About 10,000 cells for each group were then analyzed by flow cytometry as above.
Caspase 3/7 activity assay
After 0% to 2% CSE treatments, HGFs were subjected to Caspase 3/7 activities measurement with Caspase-Glo assay kit (Promega, USA). Briefly, about 10,000 cells were cultured on a 96-well plate and incubated with 0% to 2% CSE. 100 μl of Caspase-Glo reagent was added to each well and the content of well was gently mixed with a plate shaker at 200 rpm for one minute. The plate was then incubated at room temperature for 2 hours. The luminescence of each sample was measured in a microplate reader (Perkin Elmer, USA). The experiments were performed in triplicate and repeated on three separately-initiated cultures.
Immunocytochemistry
HGFs were cultured on six-well plastic plates. Following desired 0% to 2% CSE treatment, they were washed twice with PBS and then fixed with 4% formaldehyde in 0.5 ml of PBS for 30 min at room temperature. The HGFs were then washed again with PBS, blocked with PBS containing 0.5% BSA, and then incubated for 1 h with primary antibody against LC3II (Cell Signaling Technology, USA). The cells were then washed three times for 10 min per wash with PBS and incubated with a FITC-conjugated goat anti-rabbit IgG secondary antibody (Abcam, USA) for 1 h. All images were visualized by fluorescent microscopy (Zeiss, Germany) and were transferred to a computer equipped with Zen Light Edition (Zeiss, Germany) for analysis.
Statistical analysis
All experiments were performed using at least three independent cell cultures. Error bars in all figures represent the mean ± SD. The Student's two-tailed t-test was used for the determination of statistical relevance between groups, and a P value of <0.05 was considered significant.
Results
CSE induces oxidative stress and reduces human gingival fibroblast (HGF) viability
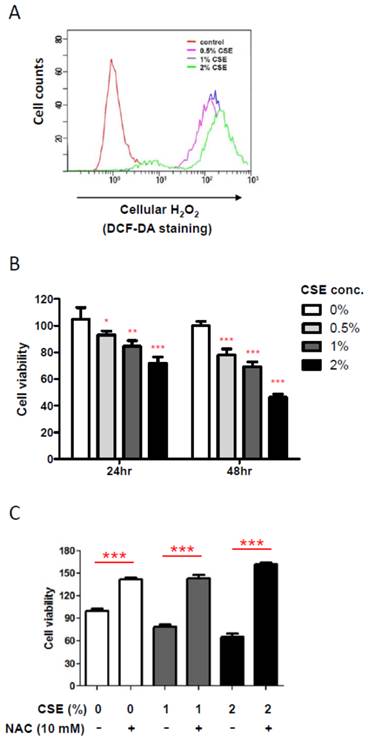
In order to obtain an insight into an effect of CSE on HGF, we first examined cellular hydrogen peroxide (H2O2) levels, an oxidative stress indicator, by flow cytometry after exposing various doses of CSE on HGFs since it has been reported that CSE induces production of cellular reactive oxygen species (ROS) in other mammalian cells. As shown in Fig. 1A, CSE exposure for 24 hr led HGFs to produce cellular H2O2 in a dose-dependent manner, indicating that CSE induces a cellular oxidative stress in HGFs. Because oxidative stress is well known to affect cell viability, we then examined changes in HGF viabilities under various CSE exposures by doses and times. From Fig. 1B, 24 hr exposure of CSE reduced HGF viabilities in a dose-dependent manner. HGF viabilities were reduced to about 20% by 1% CSE treatment and to nearly 30% by 2% CSE. In addition, HGF viabilities were further decreased in response to a longer CSE exposure (for 48 hr) when compared to the same doses for 24 hr exposures. It should be noted that these cell viability data for both 24 and 48 hr CSE treatments were obtained from a single assay in triplicates independent experiments. In order to link a CSE-induced oxidative stress and a decrease in viabilities of HGFs exposed to CSE, we used n-acetyl cysteine (NAC), an antioxidant, to reduce CSE-induced oxidative stress. As shown in Fig. 1C, when NAC was applied together with 1% or 2% CSE, HGF viabilities were similar to that of no CSE exposure, indicating that CSE-derived oxidative stress underlies the reduced viabilities of HGFs treated with CSE. Together, our results have confirmed a role for CSE in producing cellular ROS in HGFs and showed that these ROS reduced HGF viabilities in vitro.
Cigarette smoke extract (CSE) promotes cellular ROS production and reduces viability of human gingival fibroblasts (HGFs). A: Flow cytometry analysis for cellular H2O2 levels by DCF-DA staining after CSE treatments on HGFs for 24 hr. B: HGF viability followed by 24 hr and 48 hr treatments. C: HGF viability followed by 24hr CSE treatments in combination with NAC as indicated. Data are expressed as the mean ± SD, * indicates P < 0.05, ** indicates P < 0.01 and *** indicates P < 0.001.
CSE triggers apoptotic cell death in HGFs
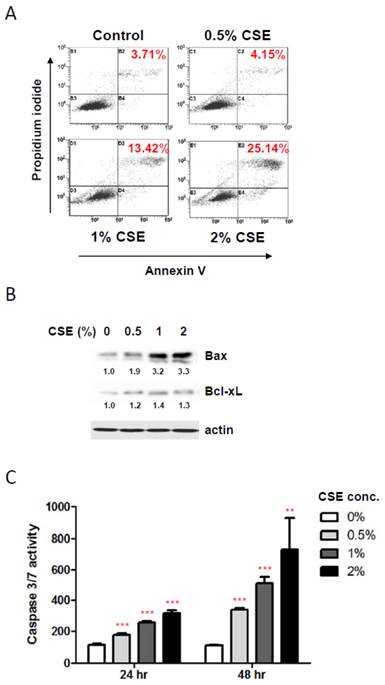
To elucidate the mechanisms underlying the reduction in HGF viability by CSE exposures, we first examined a possible induction of apoptosis in HGF cultures in response to CSE treatments using staining HGFs with Annexin V and propidium iodide followed by flow cytometry. As shown in Fig. 2A, 1%, and 2% CSE exposures (for 24 hr) resulted in an apparent apoptotic death (positive for both Annexin V and propidium iodide staining) in HGF cultures compared to control HGFs (no CSE exposure) although 0.5% CSE exposure failed to trigger a clear apoptotic cell death. In line with viability data shown in Figure 1B, live cell portions were about 82% and 68% in 1% and 2% CSE-exposed HGF cultures respectively. Next, in order to understand molecular events during CSE-induced apoptosis in HGFs, we examined pro- and anti- apoptotic protein expressions. As shown in Fig. 2B, the Bax protein, a well-known pro-apoptotic factor, was clearly induced by 1% and 2% CSE exposures (24 hr) while an anti-apoptotic protein Bcl-xL expressions were relatively similar under various CSE exposures although there was a mild increase in Bcl-xL levels in response to CSE exposures. We also examined activities of caspase-3 and caspase-7, apoptotic executors, in HGF cultures after CSE treatments. From Fig. 2C (for both 24 and 48 hr CSE exposures), caspase 3/7 activities were increased by CSE exposures dose-dependently as well as treatment-time-dependently. Similar to our cell viability assays, caspase 3/7 activity data both 24 and 48 hr CSE treatments were obtained from a single assay in triplicates of independent experiments. Overall, these results indicate that CSE promotes apoptotic death in HGF in response to CSE exposures via both up-regulation of Bax and activation of caspase 3/7.
CSE exposures induce apoptotic death in HGFs. A: Flow cytometry analysis for discrimination of HGFs using double staining with propidium iodide and FITC-labeled annexin-V after CSE treatments for 24 hr. Percentage of double positive cells (apoptotic death) were indicated in red. B: Western blot analysis for pro-apoptotic Bax and anti-apoptotic Bcl-xL protein expression levels in HGFs followed by 24hr CSE treatments. Quantitation of Bax and Bcl-xL protein levels normalized to no CSE treatment sample (0% CSE) was shown under each band. Actin was probed as a loading control. C: Caspase 3/7 activity assay for apoptosis in HGFs with 24 hr and 48 hr CSE treatments. Data are expressed as the mean ± SD, ** indicates P < 0.01 and *** indicates P < 0.001.
CSE induces autophagy in HGFs
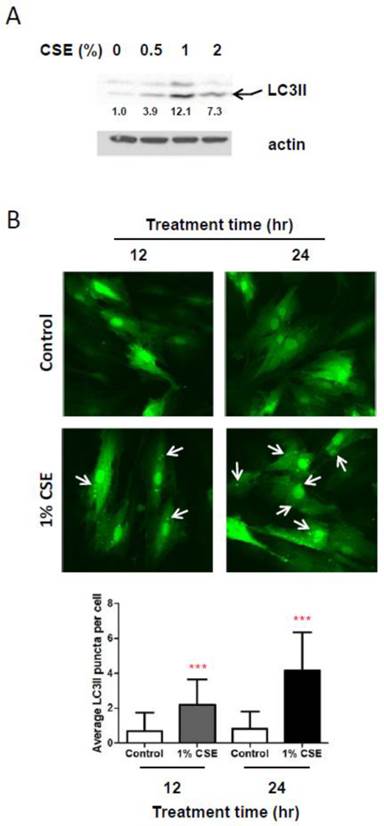
Because autophagy, a well-known process for recycling cellular components and damaged organelles, is known to occur in response to oxidative stress, we examined whether autophagy is induced in HGFs by CSE treatments. We first looked at protein levels of LC3II, a reliable marker of autophagosome by Western blotting assays. As shown in Fig. 3A, LC3II levels are clearly increased in response to CSE exposures. A slight increase in LC3II levels was observed in HGFs exposed by 0.5% while a more clear increase in LC3II levels were seen in HGFs treated with 1% or 2% CSE. Interestingly, we found that LC3II levels are higher in 1% CSE treatment than 2% CSE. We observed this trend of LC3II expression at least four independent repeats. These results indicated that HGFs trigger autophagy in response to CSE exposures. To confirm this CSE-induced autophagy in HGFs, we also monitored CSE-treated HGFs using fluorescence microscopy for LC3II fluorescent puncta in autophagosomes. From Fig. 3B, in accordance with LC3II Western blot data (Fig. 3A), cytosolic LC3II puncta were observed clearly in 1% CSE-treated HGFs at both 12 and 24 hr incubation time points. The numbers of LC3II puncta per cell were increased in 1% CSE treated HGFs compared to no CSE treatment samples (0% CSE). LC3II puncta were also seen more clearly by a longer CSE exposure time (i.e., higher in 24 hr compared to 12 hr CSE treatment). Together, the finding that both up-regulation of LC3II protein levels and presence of cytosolic fluorescence LC3II puncta were observed in HGFs treated with CSE demonstrated that autophagy occur in HGFs upon CSE exposures.
CSE exposures cause autophagy in HGFs. A: Western blot analysis for LC3II (an autophagy marker) protein expression levels in HGFs followed by 24hr CSE treatments. Quantitation of LC3II protein levels normalized to no CSE treatment sample (0% CSE) was shown under each band. Actin was probed as a loading control. B: Immunocytochemistry analysis for LC3II fluorescent puncta (cytosolic autophagosomes) in HGFs treated with CSE. Arrows indicate cytosolic LC3II puncta. Quantitation of LC3 puncta per cell was presented in the bottom margin.
Autophagy attenuates the reduction in HGF viabilities by CSE
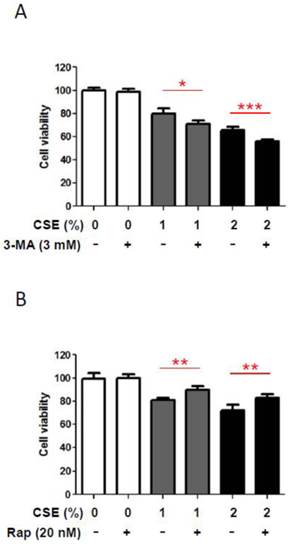
In order to understand a biological role of CSE-induced autophagy in HGFs, we inhibited CSE-induced autophagy pharmacologically and then examined whether this blockage of CSE-induced autophagy could affect HGF viabilities. We measured HGF viabilities under CSE exposures together with 3-methyladenine (3-MA), a pharmacologic inhibitor of autophagy. As shown in Fig. 4A, in the absence of CSE 3mM of 3-MA did not affect HGF viabilities. However, in the presence of CSE (both1% and 2% CSE), 3-MA reduced HGF viability further down although those reduction scales were not large but consistent. This finding that 3-MA affects viabilities of HGFs exposed to CSE implies that autophagy may have a protective role under CSE-induced stress conditions in HGFs. In the other way, we induced autophagy pharmacologically with rapamycin and then examined whether this induction of autophagy could augment HGF viabilities under CSE exposures. As shown in Fig. 4B, rapamycin (an autophagy inducer) increased HGF viabilities under both 1% and 2% CSE treatments although these increases in cell viability were quantitatively small. This observation that rapamycin-triggered autophagy enhances viabilities of CSE-treated HGFs implies a protective effect of autophagy in HGFs exposed to CSE. Together, our results from pharmacological manipulations of autophagy with 3-MA or rapamycin suggest a beneficial role for autophagy in survival of HGFs under CSE-induced cellular stress environments.
Autophage attenuates reduction in HGF viability in response to CSE. A: HGF viability followed by 24hr CSE treatments in combination with 3-methyladenine (3-MA, an autophagy inhibitor) as indicated. B: HGF viability followed by 24hr CSE treatments in combination with rapamycin (Rap, an autophagy inducer) as indicated. Data are expressed as the mean ± SD, * indicates P < 0.05, ** indicates P < 0.01 and *** indicates P < 0.001.
Discussion
Gingival fibroblasts play a key role in maintaining homeostasis of the gingival connective tissue by both synthesizing and degrading their extracellular matrix [23]. In addition, these fibroblasts have also been implicated in local immune defenses against various pathogens including periodontal bacteria. For example, because they express several immunologic receptors, bacterial stimulation of gingival fibroblasts induces the secretion of various inflammation-mediating cytokines including IL-6, IL-8, and IL-1𝛽. These observations clearly indicate that gingival fibroblasts can participate actively in immune and inflammatory responses [24,25]. Not surprisingly, these immune responses can also be affected when gingival fibroblasts are exposed to smoke. Moreover, a huge amount of evidences have implicated the apoptosis and autophagy pathways in the body immune responses [26-29]. Inappropriate induction of apoptosis appears to be critical for the pathogenesis in several immune-mediated disorders and inflammation. There is also a complex reciprocal relationship between autophagy and immunity. For example, the autophagy proteins induce and/or suppress immune and inflammatory responses while immune and inflammatory signals also act in both induction and suppression of autophagy [30].
In the present study, we investigated possible effects of cigarette smoke on human gingival fibroblasts regarding apoptosis and autophagy. Although there are many evidences for cigarette smoke-induced apoptosis, the effects of cigarette smoke on apoptotic activation still remain controversial in several cell types including human gingival fibroblasts [8,9,12-14]. For instance, Du et al [14] reported that there was a significant reduction in apoptosis and enhanced neoplastic transformation in immortalized human bronchial epithelial cells exposed to cigarette smoke. This study suggested that failure of apoptosis by cigarette smoke may enhance neoplastic transformation in these cells. Wickenden et al [8] also showed that cigarette smoke does not trigger apoptosis but induce necrosis in human alveolar epithelial type II cell lines and primary human umbilical vein endothelial cells. In fact, they reported that cigarette smoke condensate prevented staurosporine-induced apoptosis in the alveolar epithelial cells. Regarding human gingival fibroblasts, unlike our results, a previous report by Argentin et al [13] also reported that nicotine, the major ingredient of cigarette smoke, acts as an anti-apoptotic agent. However, in contrary, our results clearly demonstrated that human gingival fibroblasts exhibited apoptotic cell death in response to cigarette smoke exposures by showing a positive Annexin V and propidium iodide staining, an accumulation of pro-apoptotic Bax, and a significant increase in caspase-3/7 activities. Therefore, our findings indicate that both up-regulation of Bax and activation of caspase-3/7 may underlie cigarette smoke-induced apoptosis in the human gingival fibroblasts. One speculation on this different outcome in studies between Argentin et al [13] and us is that pharmacological effects of nicotine may differ from those of cigarette smoke since this smoke contains more complex toxic chemicals. Overall, current discrepancies in effects of cigarette smoke on apoptotic phenotype may be explained by different concentration and duration of treatment, the species and physiological status of cells, and the anatomical source of harvested cells. Thus, further studies will be of important to determine cigarette smoke-induced apoptosis for several cell types.
Autophagy acts as a protective cellular response but it is also implicated in cell death in some settings. More intriguingly, it has been proposed that autophagy might act as an adaptive mechanism in the inflammatory process although little is known about how it is involved [16]. Interestingly, recent studies have started to dissect the relationship among autophagy, apoptosis, and cigarette smoke. For example, autophagic proteins can regulate epithelial cell death in response to cigarette smoke exposure through interactions with apoptotic proteins, in particular, by activating the extrinsic apoptotic pathway [9,12]. Genetic deletion of specific autophagic proteins can inhibit emphysema development in a mouse model of chronic cigarette smoke exposure. One possible juncture between autophagy, apoptosis, and cigarette smoke may be oxidative stress that is known to occur in response to cigarette smoke exposure. Several studies showed that compounds present in cigarette smoke have been shown to lead to the production of ROS and concurrent oxidative stress [31-34]. And oxidative stress followed by the production of ROS has been shown to induce both apoptosis and autophagy [12,15]. Interestingly, Bullon P et al [16] found that stimulation of gingival fibroblasts by periodontal bacterial lipopolysaccharide increased both mitochondrial ROS and autophagy gene expression suggesting a link between autophagy and ROS in an immune response of gingival fibroblasts. Here, our results also showed that cigarette smoke induced cellular H2O2 production and autophagy in human gingival fibroblasts. Moreover, as a one step forward, we demonstrated that this autophagic response to cigarette smoke-induced oxidative stress may have a protective role in gingival fibroblast survival since induction of autophagy promotes cell survival while repression of autophagy exacerbates cell death under cigarette smoke exposures.
In summary, we here showed that cigarette smoke can induce cellular ROS in human gingival fibroblasts. This adverse effect of cigarette smoke leads to a reduction in the fibroblast viability through inducing apoptosis. Interestingly, cigarette smoke also triggers autophagy in gingival fibroblast. Regardless whether cigarette smoke directly induces autophagy or cigarette smoke-induced apoptosis triggers autophagy, this autophagic response by gingival fibroblasts seems to be beneficial to their viability. Although further study is needed to clarify the autophagic roles in relieving cigarette smoke-induced apoptotic death in human gingival fibroblasts, considering that gingival fibroblasts have a crucial role in dental health, these findings in the present study indicate that modulation of autophagy can be a novel therapeutic target in periodontal complications with cigarette smoke.
Acknowledgements
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI13C1596). This research was also supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2014R1A1A2055288 and 2015R1A5A2008833) and by the Ministry of Science, ICT & Future Planning (2014R1A1A2A16055714).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bulmanski Z, Brady M, Stoute D, Lallier TE. Cigarette smoke extract induces select matrix metalloproteinases and integrin expression in periodontal ligament fibroblasts. J Periodontol. 2012;83(6):787-796
2. Campos ML, Correa MG, Junior FH, Casati MZ, Sallum EA, Sallum AW. Cigarette smoke inhalation increases the alveolar bone loss caused by primary occlusal trauma in a rat model. J Periodontal Res. 2014;49(2):179-185
3. Rothem DE, Rothem L, Soudry M, Dahan A, Eliakim R. Nicotine modulates bone metabolism-associated gene expression in osteoblast cells. J Bone Miner Metab. 2009;27(5):555-561
4. Wang H, Liu X, Umino T, Skold CM, Zhu Y, Kohyama T, Spurzem JR, Romberger DJ, Rennard SI. Cigarette smoke inhibits human bronchial epithelial cell repair processes. Am J Respir Cell Mol Biol. 2001;25(6):772-779
5. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495-516
6. Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell. 2014;54(2):273-280
7. Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267(5203):1456-1462
8. Wickenden JA, Clarke MC, Rossi AG, Rahman I, Faux SP, Donaldson K, MacNee W. Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. Am J Respir Cell Mol Biol. 2003;29(5):562-570
9. Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880-18885
10. Csordas A, Kreutmayer S, Ploner C, Braun PR, Karlas A, Backovic A, Wick G, Bernhard D. Cigarette smoke extract induces prolonged endoplasmic reticulum stress and autophagic cell death in human umbilical vein endothelial cells. Cardiovasc Res. 2011;92(1):141-148
11. Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, Kawaishi M, Hirano J, Odaka M, Morikawa T, Nishimura S, Nakayama K, Kuwano K. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1(5):630-641
12. Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, Cervo M, Yao H, Chung AL, Mizumura K, An CH, Shan B, Franks JM, Haley KJ, Owen CA, Tesfaigzi Y, Washko GR, Quackenbush J, Silverman EK, Rahman I, Kim HP, Mahmood A, Biswal SS, Ryter SW, Choi AM. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212-5230
13. Argentin G, Cicchetti R. Genotoxic and antiapoptotic effect of nicotine on human gingival fibroblasts. Toxicol Sci. 2004;79(1):75-81
14. Du H, Sun J, Chen Z, Nie J, Tong J, Li J. Cigarette smoke-induced failure of apoptosis resulting in enhanced neoplastic transformation in human bronchial epithelial cells. J Toxicol Environ Health A. 2012;75(12):707-20
15. Gannon AM, Stampfli MR, Foster WG. Cigarette smoke exposure elicits increased autophagy and dysregulation of mitochondrial dynamics in murine granulosa cells. Biol Reprod. 2013;88(3):63
16. Bullon P, Cordero MD, Quiles JL, Ramirez-Tortosa Mdel C, Gonzalez-Alonso A, Alfonsi S, García-Marín R, de Miguel M, Battino M. Autophagy in periodontitis patients and gingival fibroblasts: unraveling the link between chronic diseases and inflammation. BMC Med. 2012;10:122
17. Cho TJ, Wee SW, Woo VH, Choi JI, Kim SJ, Shin HI. et al. Porphyromonas gingivalis-induced autophagy suppresses cell proliferation through G1 arrest in oral cancer cells. Arch Oral Biol. 2014;59(4):370-8
18. Yu Jh, Liu Cy, Zheng Gb, Zhang LY, Yan Mh, Zhang Wy, Meng Xy, Yu Xf. Pseudolaric acid B induced cell cycle arrest, autophagy and senescence in murine fibrosarcoma l929 cell. Int J Med Sci. 2013;10(6):707-718
19. Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91(2):142-9
20. Walter C, Kaye EK, Dietrich T. Active and passive smoking: assessment issues in periodontal research. Periodontol 2000. 2012;58(1):84-92
21. Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89(12):1349-63
22. Mahanonda R, Sa-Ard-Iam N, Montreekachon P, Pimkhaokham A, Yongvanichit K, Fukuda MM. et al. IL-8 and IDO expression by human gingival fibroblasts via TLRs. J Immunol. 2007;178(2):1151-7
23. Hinz B. Matrix mechanics and regulation of the fibroblast phenotype. Periodontol 2000. 2013;63(1):14-28
24. Minami T, Kuroishi T, Ozawa A, Shimauchi H, Endo Y, Sugawara S. Histamine amplifies immune response of gingival fibroblasts. J Dent Res. 2007;86(11):1083-1088
25. Liu J, Du X, Chen J, Hu L, Chen L. The induction expression of human β-defensins in gingival epithelial cells and fibroblasts. Arch Oral Biol. 2013;58(10):1415-1421
26. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883-899
27. Ryter SW, Lee SJ, Choi AM. Autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. Expert Rev Respir Med. 2010;4(5):573-584
28. Verschuere S, Bracke KR, Demoor T, Plantinga M, Verbrugghe P, Ferdinande L, Lambrecht BN, Brusselle GG, Cuvelier CA. Cigarette smoking alters epithelial apoptosis and immune composition in murine GALT. Lab Invest. 2011;91(7):1056-67
29. Wu Y, Zhang Y, Wang L, Diao Z, Liu W. The Role of Autophagy in Kidney Inflammatory Injury via the NF-κB Route Induced by LPS. Int J Med Sci. 2015;12(8):655-667
30. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323-335
31. Barua RS, Ambrose JA, Srivastava S, DeVoe MC, Eales-Reynolds LJ. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: an in vitro demonstration in human coronary artery endothelial cells. Circulation. 2003;107(18):2342-2347
32. Valavanidis A, Vlachogianni T, Fiotakis K. Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int J Environ Res Public Health. 2009;6(2):445-462
33. Cano M, Thimmalappula R, Fujihara M, Nagai N, Sporn M, Wang AL, Neufeld AH, Biswal S, Handa JT. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vision Res. 2010;50(7):652-664
34. Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. 2011;254(2):72-85
Author contact
![]() Corresponding authors: June-Ho Byun (Department of Oral and Maxillofacial Surgery, Gyeongsang National University School of Medicine and Gyeongsang National University Hospital, Institute of Health Sciences, Gyeongsang National University, Chilam-dong, Jinju, 660-702, Republic of Korea, Tel : 82-55-750-8258, Fax : 82-55-761-7024, E-mail address : surbyunac.kr) or Dong Kyun Woo (College of Pharmacy and Research Institute of Pharmaceutical Sciences, Gyeongsang National University, Jinju, Republic of Korea, Tel : 82-55-772-2428, E-mail address : dongkyun.wooac.kr ).
Corresponding authors: June-Ho Byun (Department of Oral and Maxillofacial Surgery, Gyeongsang National University School of Medicine and Gyeongsang National University Hospital, Institute of Health Sciences, Gyeongsang National University, Chilam-dong, Jinju, 660-702, Republic of Korea, Tel : 82-55-750-8258, Fax : 82-55-761-7024, E-mail address : surbyunac.kr) or Dong Kyun Woo (College of Pharmacy and Research Institute of Pharmaceutical Sciences, Gyeongsang National University, Jinju, Republic of Korea, Tel : 82-55-772-2428, E-mail address : dongkyun.wooac.kr ).