Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(2):92-98. doi:10.7150/ijms.13785 This issue Cite
Research Paper
Gelsolin and Progression of Aortic Arch Calcification in Chronic Hemodialysis Patients
Terry Ting-Yu Chiou1, Shang-Chih Liao1, Yu-Yin Kao1, Wen-Chin Lee1, Yueh-Ting Lee1, Hwee-Yeong Ng1, Po-Shun Lee2, Chien-Te Lee1 ![]()
1. Division of Nephrology, Department of Internal Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang-Gung University College of Medicine, Taiwan.
2. Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA.
Received 2015-9-7; Accepted 2016-1-5; Published 2016-1-29
Abstract
Background: Vascular calcification (VC) is a key process associated with cardiovascular mortality in dialysis patients. Gelsolin is an actin-binding protein that can modulate inflammation, correlated inversely with hemodialysis (HD) mortality and involved in bone calcification homeostasis. In this report, we aim to characterize progression in aortic arch calcification (AAC) and investigate its association with gelsolin.
Methods: 184 HD patients were enrolled and their annual posterior-anterior chest X-ray films (CXR) in 2009 and 2013 were examined. The severity of AAC was classified as grade 0 to 3. Blood levels of gelsolin were measured by ELISA kits. Biographic and biochemical data at baseline were analyzed with status of AAC at baseline and changes after 4 years.
Results: At baseline, 60% of the patients had detectable AAC on CXR. After 4 years, 77% had AAC. Patients with grade 1 and 2 AAC had increased risk of progression (Odds ratio [OR] 2~3, P=0.001) compared to those with grade 0 at baseline. Compared to those with no AAC, patients with AAC progression had older age, lower gelsolin, higher waist circumference and prevalence of vascular disease. Regression analysis confirmed baseline gelsolin (odds ratio 0.845, 95% confidence interval [0.734-0.974]) and waist circumference as the independent factors associated with AAC progression. Gelsolin is positively correlated with serum albumin and negatively with tumor necrosis factor-alpha.
Conclusion: Our study demonstrated that HD patients with grades 1 or 2 baseline AAC are at increased risk of further progression compared to those with grade 0. We also found lower blood levels of gelsolin associated with progressive AAC. Further investigation into the mechanistic roles of gelsolin in vascular calcification may provide new understanding of this key process.
Keywords: Aortic arch calcification, gelsolin, hemodialysis.
Introduction
Cardiovascular disease is the most common cause of death in patients on chronic dialysis [1]. Vascular calcification (VC) is increasingly recognized as a key process contributing to the high cardiovascular mortality in dialysis patients [2-4]. Our previous study on 712 prevalent hemodialysis (HD) patients showed a strong correlation between aortic arch calcification (AAC) and 10-year mortality [5]. It also substantiated the use of plain chest X-ray films (CXR) as a simple tool to evaluate AAC.
Recent studies reported that 26~78% of dialysis patients had various degrees of AAC on CXR, and 34~60% had progression after 1~5 years of follow-up [6-10]. These variations in the prevalence of calcification and its progression may be related to the differences in ethnicity, comorbidity, length of observation, and methods of assessment. Progression in VC has also been associated with mortality [6,7]. Factors contributing to VC include age, comorbidity (diabetes, hypertension, metabolic syndrome, dyslipidemia), dialysis vintage, medications (calcium, vitamin D, coumadin), and uremia-related mineral bone disorder (serum calcium [Ca], phosphorus [P], parathyroid hormone [PTH]) [11-15]. Markers of inflammation and oxidative stress have also been implicated [16,17].
Gelsolin, by regulating intracellular actin filaments, is important in cell morphology, migration and phagocytosis [18]. Its extracellular isoform, plasma gelsolin is secreted by different types of cells and serves as a scavenging system for potentially toxic actin filaments [19]. It can localize inflammation and minimize tissue damage by binding excessive actins released from tissue injury. The phenomenon of plasma gelsolin depletion has been observed in different diseases. Specifically, in patients with acute myocardial infarction and fulminant hepatic failure, plasma gelsolin levels dropped dramatically and recovered if organ function improved from injury [20]. Further research found that in addition to actin, plasma gelsolin may also modulate immune response by binding to key inflammatory mediators, including lipopolysaccharide, lysophospholipids, and platelet activating factor [21,22].
Furthermore, gelsolin interacts with osteopontin and both are involved in bone homeostasis [23]. In HD patients, plasma gelsolin levels were reduced nearly 50% compared to healthy volunteers and correlated with mortality and protein-energy wasting [24,25]. However, whether gelsolin have independent roles in vascular calcification is uncertain, and strategies to reduce inflammation and protect vascular integrity are poorly defined. In this report, we aim to characterize the progression of aortic arch calcification in HD patients and investigate its associations with novel biomarker gelsolin.
Subjects and Methods
From January to December 2009, we enrolled 184 stable HD patients from Kaohsiung Chang Gung Memorial Hospital. Inclusion criteria were over 20 years of age and regular 4-hour HD session three times a week for at least 6 months. Exclusion criteria were chronic viral hepatitis, malignancy (within 5 years), acute infection or hospitalization (within three months). This study was approved by the Institutional Review Boards and Ethics Committee in Chang Gung Memorial Hospital (IRB No. 98-2685B), and informed consents were obtained from all participants.
Laboratory and Clinical Data
For every study participant, blood samples were collected. The averages of biochemical data were obtained over the three months prior to study enrollment. The adequacy of dialysis was assessed by Kt/V urea using the urea kinetic model of Gotch [26]. Gelsolin levels in blood were measured with ELISA kits from Critical Biologics Corporation (Cambridge, MA. USA). The immunoassay kits for interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) were from R&D Systems, Minneapolis, MN. USA. All measurements were done in duplicate. All patients' medical records were carefully reviewed. Vascular disease was identified by documented history of ischemic heart disease, cerebrovascular disease or peripheral vascular disease.
Evaluation of AAC on Chest Radiography
Our HD patients have chest X-ray examinations (posterior-anterior approach, standing) annually. For each study participant, the routine annual chest X-ray film in 2009 was selected as the baseline. A second follow-up chest X-ray film was selected in 2013 or the year when the patient reached outcomes such as death, kidney transplantation or transfer to other dialysis centers. A simple classification by Symeonidis et al. [27] was used to evaluate AAC in the present study. Briefly, the severity of calcification was classified as grade (Gr.) 0 (no calcification visible), Gr. 1 (single thin or small spots of calcification), Gr. 2 (one or more areas of thick calcification, but ≤ 50% of the circular area of the aortic knob) and Gr. 3 (circular calcification with >50% of circular area of the aortic knob).
Each posterior-anterior (P-A) chest X-ray (CXR) of these HD patients was read and graded independently by two readers (one nephrologist and one trained nurse practitioner) blinded to the patients' clinical data. Eight X-rays were graded differently on calcification score (±1). The discrepancies between the two observers were resolved by a third independent reader. The progression of aortic calcification was determined by comparing the grades at baseline and at follow-up. “Progressive AAC” was defined if the second chest X-ray's grade is greater than the first one. “Stable AAC” was defined if the first CXR showed calcification and the second CXR had the same calcification grade. “No AAC” group was defined if both the first and second CXR's were grade 0.
Statistical Analysis
Statistical analysis was performed using SPSS version 12.0. Results were expressed as mean ± standard deviation or median (interquartile range) for nonparametric data. Comparisons between two groups were performed using Student t-test or Mann-Whitney test for nonparametric data. We used multiple logistic regressions to examine the independent relationships between AAC progression (progressive vs. no AAC) and other variables. Regression adjusted for age, HD vintage, vascular disease, BMI, waist circumference, Kt/V, parameters of mineral-bone disorder (serum calcium [Ca], phosphorus [P] and intact parathyroid hormone [iPTH]), gelsolin and IL-6. All results were considered significant if P-value was less than 0.05.
Results
Aortic arch calcification and its progression
At baseline, the subjects with and without AAC had similar dialysis adequacy (Kt/V 1.38 ± 0.22 vs. 1.45 ± 0.23, P>0.05) and biochemical control, particularly serum calcium (9.2 ± 0.8 vs. 9.4 ± 0.9 mg/dL, P>0.05) and phosphate (4.8 ± 1.4 vs. 4.9 ± 1.4 mg/dL, P>0.05). 75 (40%) patients had no detectable AAC (grade 0) on plain chest X-ray. 52 (28%) patients had grade 1, 43 (23%) grade 2, and 14 (8%) grade 3 (Figure 1A).
Distribution of different grades of aortic arch calcification (AAC) at (A) baseline and (B) after 4 years of follow-up.
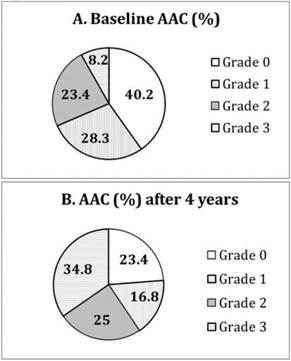
After 4 years of follow-up, only 43 (23%) patients remained at grade 0. Thirty-one (17%) patients had grade 1, 46 (25%) grade 2, and 64 (35%) grade 3 (Figure 1B). Moreover, 32 (43%) of those with grade 0 at baseline progressed to higher grades of AAC over 4 years. 73% (38/52) of those with grade 1 at baseline had progressive AAC, and 84% (36/43) of those with grade 2 at baseline had progressive AAC. Fourteen patients with grade 3 calcification at baseline remained so after 4 years. Compared to those with grade 0 (the reference group), patients with grade 1 at baseline had increased risk of further progression in AAC (Odds ratio [OR] 2.11, 95% confidence interval [CI] 1.29-3.44, P=0.001, adjusted for age, waist circumference, gelsolin and IL-6). Patients with grade 2 at baseline also had higher risk of progression in AAC (OR 3.49 [1.72-7.07], P<0.001).
Factors associated with aortic arch calcification and its progression
We compared the clinical and biochemical profiles between patients with and without AAC. At baseline, patients with calcifications had significantly lower serum albumin (3.9 ± 0.3 vs. 4.0 ± 0.2 g/dL, P=0.015). They also tend to have older age (61 ± 10 vs. 58 ± 12, P=0.066) and lower gelsolin (824 ± 257 vs. 898 ± 272 µU/ml, P=0.064). However, the two groups did not differ significantly in serum Ca, P, iPTH, cholesterol or high-sensitivity C-reactive protein (hs-CRP).
At 4-year follow-up, patients with progressive AAC had significantly older age, higher waist circumference, IL-6, and lower gelsolin than those with no AAC (Table 1). They also had significantly higher rate of vascular disease (51% vs. 31%, P<0.05). There were no significant differences between patients with stable and progressive AAC. Multiple logistic regression (adjusting for age, HD vintage, vascular disease, BMI, waist circumference, Kt/V, serum calcium, phosphorus, iPTH, gelsolin and IL-6) confirmed baseline gelsolin and waist circumference as the independent factors associated with progressive AAC (Table 2). For every 10cm increase in waist circumference, the risk of AAC progression is increased by 47%. For every 100μU/ml increase in gelsolin level, the risk of AAC progression is decreased by 15%.
Comparison between 3 groups of patients (no AAC, stable AAC and progressive AAC) after 4 years of follow-up.
| No AAC (N = 43) | Stable AAC (N = 35) | Progressive AAC (N=106) | |
|---|---|---|---|
| Age, year | 56 ± 12 | 61 ± 8* | 60 ± 10** |
| HD vintage, month | 52 (28-113) | 68 (37-121) | 62.0 (36.0-102.0) |
| BMI, kg/m² | 22.1 ± 4.5 | 23.5 ± 4.1 | 22.8 ± 3.5 |
| Waist circumference, cm | 81.2 ± 12.1 | 86.7 ± 13.7 | 85.7 ± 11.7** |
| Vascular disease | 31% | 46% | 51%** |
| Kt/V | 1.4 ± 0.2 | 1.4 ± 0.2 | 1.4 ± 0.2 |
| nPCR, g/kg/day | 1.22 ± 0.3 | 1.18 ± 0.31 | 1.19 ± 0.28 |
| Hemoglobin, g/dL | 10.4 ± 1.0 | 10.5 ± 1.5 | 10.6 ± 1.2 |
| Serum albumin, g/dL | 4.0 ± 0.2 | 3.9 ± 0.2 | 3.9 ± 0.3 |
| BUN, mmol/L | 69.7 ± 10.2 | 70.8 ± 15.0 | 68.6 ± 15.1 |
| Serum creatinine, mg/dL | 10.8 ± 1.9 | 11.2 ± 2.1 | 10.8 ± 2.1 |
| Calcium, mg/dL | 9.2 ± 0.8 | 9.1 ± 0.9 | 9.4 ± 0.8 |
| Phosphorus, mg/dL | 4.8 ± 1.5 | 4.9 ± 1.3 | 4.9 ± 1.4 |
| iPTH, pg/dL | 259 (98-687) | 231 (110-542) | 238 (86-627) |
| Total cholesterol, mg/dL | 183.6 ± 40.7 | 189.7 ± 38.3 | 188.2 ± 39.3 |
| Triglyceride, mg/dL | 164.0 ± 105.7 | 144.7 ± 94.8 | 159.4 ± 106.5 |
| HDL, mg/dL | 46.6 ± 12.9 | 54.2 ± 16.3 | 45.1 ± 14.9 |
| LDL, mg/dL | 101.0 ± 42.2 | 110.4 ± 23.3 | 110.2 ± 34.2 |
| hsCRP, mg/L | 2.3 (1.0-5.1) | 3.1 (1.5-6.8) | 2.9 (1.3-7.4) |
| Gelsolin, µU/ml | 925.5 ± 277.4 | 846.9 ± 235.7 | 827.6 ± 266.8** |
| TNF-α, pg/mL | 5.7 (4.1-8.5) | 7.1 (4.1-9.7) | 6.7 (3.9-11.3) |
| IL-6, pg/mL | 2.0 (1.4-3.8) | 3.0 (1.7-6.4)* | 2.7 (1.5-4.5)** |
“No AAC” means no detectable aortic arch calcification on CXR at baseline and at follow-up. “Stable AAC” means same AAC on CXR at baseline and at follow-up.
“Progressive AAC” means AAC grades at follow-up higher than at baseline.
* P<0.05 Stable AAC vs. No AAC
** P<0.05 Progressive AAC vs. No AAC
Abbreviations BMI: body mass index; nPCR: normalized protein catabolic rate; iPTH: intact parathyroid hormone; HDL: high density lipoprotein cholesterol; LDL: low density lipoprotein cholesterol; hsCRP: high-sensitivity C-reactive protein; TNF-α: tumor necrosis factor-alpha; IL-6: interleukin-6.
Multiple logistic regression* analysis showing the independent factors associated with progressive aortic arch calcification after 4 years of follow-up.
| Odds Ratio (95%CI) | P-value | |
|---|---|---|
| Age (per 1-year increase) | 1.022 (0.985-1.062) | NS |
| HD vintage (per 1 year increase) | 1.007 (0.999-1.015) | NS |
| Vascular disease (yes vs. no) | 1.348 (0.586-3.100) | NS |
| BMI (per 1kg/m2 increase) | 0.892 (0.756-1.008) | NS |
| Waist circumference (per 10cm increase) | 1.475 (1.050-2.074) | 0.025 |
| Kt/V (per 1 unit increase) | 0.601 (0.127-3.452) | NS |
| Calcium (per 1 mg/dL increase) | 1.008 (0.621-1.636) | NS |
| Phosphorus (per 1 mg/dL increase) | 0.976 (0.973-1.238) | NS |
| iPTH (per 1 pg/dL increase) | 1.000 (0.999-1.001) | NS |
| Gelsolin (per 100μU/ml increase) | 0.845 (0.734-0.974) | 0.02 |
| IL-6 (per 1 pg/mL increase) | 1.116 (0.986-1.262) | NS |
*Regression (progressive vs. no AAC) adjusted for age, HD vintage, vascular disease, BMI, waist circumference, Kt/V, serum calcium, phosphorus, iPTH, gelsolin and IL-6.
Abbreviations: CI, Confidence Interval; BMI, body mass index; BMI: body mass index; iPTH: intact parathyroid hormone; IL-6: interleukin-6; NS, non-significant.
We also analyzed the associations between gelsolin and important factors. Baseline gelsolin is positively correlated with serum albumin (ρ [Pearson's coefficient] 0.262, P<0.001) and creatinine (ρ 0.276, P<0.001), and negatively correlated with age (ρ -0.212, P=0.004), TNF-α (ρ -0.148, P=0.044) and white blood cell counts (ρ -0.163, P=0.026).
Discussion
This study on 184 prevalent HD patients had two major findings. First, the severity of AAC on plain chest X-ray confers different risks for AAC progression and vascular disease. Patients with higher grade AAC at baseline had higher risk of progression, and subjects with AAC progression have higher rate of vascular disease. Second, baseline gelsolin level was independently associated with progression in AAC. Other important factors of AAC and progression were serum albumin and waist circumference.
Our study demonstrated that AAC is common in HD patients, 60% at baseline and 77% after 4 years of follow-up. In addition, we showed that progression in AAC is associated with higher prevalence of vascular disease. Sigrist et al. [12], using multislice computed tomography to quantify calcification in superficial femoral artery reported a constant progression rate over the first and second years of observation. Their study examined a heterogeneous group of 101 patients with stage 4 CKD, HD and peritoneal dialysis. However, from our cohort of HD patients, we found that progression in AAC over a longer time period (4 years) is dependent on the initial calcification status. Less than half of those with no evident AAC on baseline CXR developed calcification over 4 years of follow-up. However, 73% of those with baseline grade 1 AAC and 84% of those with grade 2 had more severe calcification over the same period. This observation suggests that when AAC becomes evident on CXR, the risk of progression is much higher. This has several important clinical implications. First, the progression of AAC may not be the same for everyone over 4 years, which makes risk stratification and timing of intervention extremely crucial: the later stage we intervene, the less time we may have to deliver an effective therapy. Second, routine assessment of AAC will help identify patients at increased risk of progression in vascular calcification. Third, plain chest X-ray is an easily attainable yet powerful tool in the assessment of vascular calcification and its risk of progression.
The independent association between plasma gelsolin and progression in aortic arch calcification has not been reported in the literature before. Gelsolin is involved in bone homeostasis through interaction with osteopontin, an inhibitory protein in vascular calcification. In osteoclast, osteopontin binds to integrin αvβ3 and stimulates gelsolin-associated polyphosphoinositides, leading to alterations in cytoskeleton and bone resorption [28]. Response to osteopontin stimulation was absent in gelsolin-deficient osteoclast [23]. In gelsolin-deficient adult mice, bone resorption was diminished, resulting in increased bone mass. Gelsolin, together with osteopontin promotes resorption, and modifies the process of calcification. In patients with chronic kidney disease (CKD), the diminishing gelsolin levels may contribute to the imbalance between the activators and inhibitors of vascular calcification. In normal bone homeostasis, osteoblasts promote the recruitment and activation of osteoclasts, resulting in bone resorption and remodeling. However, in vascular calcification, the trans-differentiated vascular smooth muscle cells with an osteoblast phenotype do not promote resorption of calcification by osteoclast-like cells [29]. In patients with CKD, a gelsolin depleted state may promote the propagation of calcification by exacerbating the osteoclast-like cells' inability to resorb hydroxyapatite crystals. It is tempting to speculate that correction of gelsolin deficiency may reactivate osteoclasts, leading to resorption of vascular calcification.
Our findings also complement previous work demonstrating the connections between gelsolin, atherosclerosis, inflammation and oxidative stress, which are also important mediators of vascular injury and calcification (Figure 2).
Possible mechanisms for gelsolin depletion, vascular injury and calcification in chronic kidney disease. Gelsolin depletion may impair modulation on lysophospholipids, macrophage receptor and nitric oxide system which are involved in the maintenance of vascular integrity, development of foam cells, plaque and atherosclerosis. Blood levels of gelsolin have also been correlated inversely with oxidative stress and inflammatory cytokines (i.e. TNF-α). In addition to an impaired resorption of calcification by osteoclast-like cells, gelsolin depletion may also exacerbate the imbalance between calcification inhibitors and activators.
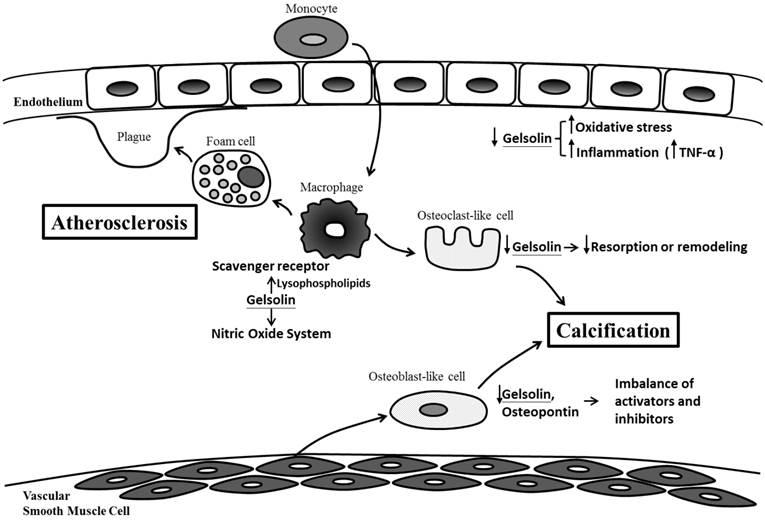
Macrophage is a key player in the process of inflammation and atherosclerosis. Gelsolin can alter macrophage function by triggering nitric oxide synthase, leading to reduced inflammation [30]. In animal models of sepsis, supplementation with recombinant human plasma gelsolin (rhu-pGSN) to restore its depleted level can provide favorable inflammatory modulation and survival benefits [31]. Gelsolin can also modulate cellular responses to lysophospholipids which interact with macrophage receptors and mediate atherosclerosis [21,32]. Indeed, gelsolin expression was reduced in the media layer of human atherosclerotic coronary arteries [33], and its secretion into the ex vivo culture medium was also decreased [34]. This depletion of gelsolin may have consequences in vascular functions. In atherosclerosis and CKD, depletion of gelsolin may impair ligand interaction with macrophage receptors, the modulation on nitric oxide system, and the buffering capacity for inflammatory mediators. Ours and other studies also support the connections between alterations in gelsolin levels and inflammation, oxidative stress, and production of reactive oxygen species [35-37]. In addition to its role as a novel biomarker, gelsolin has therapeutic potential. In mouse models of sepsis and multiple sclerosis where gelsolin levels were depleted, administration of recombinant gelsolin proteins improved survival and brain inflammation [31,40]. In a recent preliminary clinical trial, the safety of rhu-pGSN was established in humans [38].
In addition to the characterization of AAC and its progression over 4 years, our study also explored the important factors associated with these phenomena. In our study, markers of mineral bone metabolism (Ca, P, iPTH) were not associated with AAC and its progression. This is likely because our patients were in good control from this perspective. However, despite keeping their serum Ca, P, PTH within the guideline recommendations, these patients still have high rate of AAC and progression. It should be emphasized that other important factors must be involved. Gelsolin, the novel protein may be one of these key players in AAC. Our analysis also showed that lower serum albumin is associated with baseline AAC. Moreover, higher waist circumference is associated with AAC progression. Increased waist circumference suggests increased abdominal adiposity, which is also associated with coronary artery calcification in CKD patients [39].
Our study has several limitations. First, using plain CXR and 4-grade scoring method to evaluate the severity of vascular calcification may underestimate or miss some subtle calcification and changes. CT scan would offer a much higher resolution, a finer differentiation of calcification, but more costly. Secondly, our study does not have detailed accounts of patients' medications because they may have various sources of medications outside of our dialysis center. Lastly, our study results may not be generalizable to patients of different ethnicity or other modality of renal replacement therapy.
In conclusion, our study found that AAC based on plain CXR was common in HD patients. Patients with initially higher grades of AAC are at increased risk of further progression, which is associated with higher risk of vascular disease. Our identification of plasma gelsolin as a novel protein associated with progression in VC warrants further confirmation in larger population and may offer new understanding and therapeutic strategies toward vascular injuries and calcification.
Acknowledgements
Grant support by Kaohsiung Chang Gung Memorial Hospital (CMRPG8B0491) and Minister of Science and Technology, Republic of China (MOST 104-2314-B-182A- 143).
Competing Interests
None to declare.
References
1. Collins AJ, Li S, Ma JZ, Herzog C. Cardiovascular disease in end-stage renal disease patients. Am J Kidney Dis. 2001;38:S26-29
2. McIntyre CW. The functional cardiovascular consequences of vascular calcification. Semin Dial. 2007;20:122-128
3. Cannata-Andia JB, Rodriguez-Garcia M, Carrillo-Lopez N, Naves-Diaz M, Diaz-Lopez B. Vascular calcifications: Pathogenesis, management, and impact on clinical outcomes. J Am Soc Nephrol. 2006;17:S267-273
4. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vasc Health Risk Manag. 2009;5:185-197
5. Lee CT, Huang CC, Hsu CY, Chiou TT, Ng HY, Wu CH, Kuo WH, Lee YT. Calcification of the aortic arch predicts cardiovascular and all-cause mortality in chronic hemodialysis patients. Cardiorenal Med. 2014;4:34-42
6. Goldsmith DJ, Covic A, Sambrook PA, Ackrill P. Vascular calcification in long-term haemodialysis patients in a single unit: A retrospective analysis. Nephron. 1997;77:37-43
7. Ammirati AL, Dalboni MA, Cendoroglo M, Draibe SA, Santos RD, Miname M, Canziani ME. The progression and impact of vascular calcification in peritoneal dialysis patients. Perit Dial Int. 2007;27:340-346
8. Kim HG, Song SW, Kim TY, Kim YO. Risk factors for progression of aortic arch calcification in patients on maintenance hemodialysis and peritoneal dialysis. Hemodial Int. 2011;15:460-467
9. Ogawa T, Ishida H, Akamatsu M, Matsuda N, Fujiu A, Ito K, Ando Y, Nitta K. Relation of oral 1alpha-hydroxy vitamin d3 to the progression of aortic arch calcification in hemodialysis patients. Heart Vessels. 2010;25:1-6
10. Tamei N, Ogawa T, Ishida H, Ando Y, Nitta K. Serum fibroblast growth factor-23 levels and progression of aortic arch calcification in non-diabetic patients on chronic hemodialysis. J Atheroscler Thromb. 2011;18:217-223
11. Shroff R, Long DA, Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol. 2013;24:179-189
12. Sigrist MK, Taal MW, Bungay P, McIntyre CW. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin J Am Soc Nephrol. 2007;2:1241-1248
13. Noordzij M, Cranenburg EM, Engelsman LF, Hermans MM, Boeschoten EW, Brandenburg VM, Bos WJ, Kooman JP, Dekker FW, Ketteler M, Schurgers LJ, Krediet RT, Korevaar JC, Group NS. Progression of aortic calcification is associated with disorders of mineral metabolism and mortality in chronic dialysis patients. Nephrol Dial Transplant. 2011;26:1662-1669
14. Iribarren C, Sidney S, Sternfeld B, Browner WS. Calcification of the aortic arch: Risk factors and association with coronary heart disease, stroke, and peripheral vascular disease. JAMA. 2000;283:2810-2815
15. Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension. 2001;38:938-942
16. Chen NX, Moe SM. Vascular calcification: Pathophysiology and risk factors. Curr Hypertens Rep. 2012;14:228-237
17. Lee CT, Chua S, Hsu CY, Tsai YC, Ng HY, Kuo CC, Wu CH, Chen TC, Chiu TT, Lee YT. Biomarkers associated with vascular and valvular calcification in chronic hemodialysis patients. Dis Markers. 2013;34:229-235
18. Bucki R, Levental I, Kulakowska A, Janmey PA. Plasma gelsolin: Function, prognostic value, and potential therapeutic use. Curr Protein Pept Sci. 2008;9:541-551
19. Lee WM, Galbraith RM. The extracellular actin-scavenger system and actin toxicity. N Engl J Med. 1992;326:1335-1341
20. Suhler E, Lin W, Yin HL, Lee WM. Decreased plasma gelsolin concentrations in acute liver failure, myocardial infarction, septic shock, and myonecrosis. Crit Care Med. 1997;25:594-598
21. Osborn TM, Dahlgren C, Hartwig JH, Stossel TP. Modifications of cellular responses to lysophosphatidic acid and platelet-activating factor by plasma gelsolin. Am J Physiol Cell Physiol. 2007;292:C1323-1330
22. Bucki R, Kulakowska A, Byfield FJ, Zendzian-Piotrowska M, Baranowski M, Marzec M, Winer JP, Ciccarelli NJ, Gorski J, Drozdowski W, Bittman R, Janmey PA. Plasma gelsolin modulates cellular response to sphingosine 1-phosphate. Am J Physiol Cell Physiol. 2010;299:C1516-1523
23. Chellaiah M, Kizer N, Silva M, Alvarez U, Kwiatkowski D, Hruska KA. Gelsolin deficiency blocks podosome assembly and produces increased bone mass and strength. J Cell Biol. 2000;148:665-678
24. Lee PS, Sampath K, Karumanchi SA, Tamez H, Bhan I, Isakova T, Gutierrez OM, Wolf M, Chang Y, Stossel TP, Thadhani R. Plasma gelsolin and circulating actin correlate with hemodialysis mortality. J Am Soc Nephrol. 2009;20:1140-1148
25. Chiu TT, Liao SC, Lee WC, Lee PS, Ng HY, Chien YS, Lee CT. Gelsolin and adipokines are associated with protein-energy wasting in hemodialysis patients. Artif Organs. 2015;39:150-155
26. Gotch FA, Sargent JA. A mechanistic analysis of the national cooperative dialysis study (NCDS). Kidney Int. 1985;28:526-534
27. Symeonidis G, Papanas N, Giannakis I, Mavridis G, Lakasas G, Kyriakidis G, Artopoulos I. Gravity of aortic arch calcification as evaluated in adult greek patients. Int Angiol. 2002;21:233-236
28. Chellaiah M, Hruska K. Osteopontin stimulates gelsolin-associated phosphoinositide levels and phosphatidylinositol triphosphate-hydroxyl kinase. Mol Biol Cell. 1996;7:743-753
29. Persy V, D'Haese P. Vascular calcification and bone disease: The calcification paradox. Trends Mol Med. 2009;15:405-416
30. Yang Z, Chiou TT, Stossel TP, Kobzik L. Plasma gelsolin improves lung host defense against pneumonia by enhancing macrophage nos3 function. Am J Physiol Lung Cell Mol Physiol. 2015;309:L11-16
31. Lee PS, Waxman AB, Cotich KL, Chung SW, Perrella MA, Stossel TP. Plasma gelsolin is a marker and therapeutic agent in animal sepsis. Crit Care Med. 2007;35:849-855
32. Nishikawa M, Kurano M, Ikeda H, Aoki J, Yatomi Y. Lysophosphatidylserine has bilateral effects on macrophages in the pathogenesis of atherosclerosis. J Atheroscler Thromb. 2015;22:518-526
33. de la Cuesta F, Zubiri I, Maroto AS, Posada M, Padial LR, Vivanco F, Alvarez-Llamas G, Barderas MG. Deregulation of smooth muscle cell cytoskeleton within the human atherosclerotic coronary media layer. J Proteomics. 2013;82:155-165
34. de la Cuesta F, Barderas MG, Calvo E, Zubiri I, Maroto AS, Darde VM, Martin-Rojas T, Gil-Dones F, Posada-Ayala M, Tejerina T, Lopez JA, Vivanco F, Alvarez-Llamas G. Secretome analysis of atherosclerotic and non-atherosclerotic arteries reveals dynamic extracellular remodeling during pathogenesis. J Proteomics. 2012;75:2960-2971
35. Genre F, Lopez-Mejias R, Miranda-Filloy JA, Ubilla B, Carnero-Lopez B, Gomez-Acebo I, Blanco R, Ochoa R, Rueda-Gotor J, Gonzalez-Juanatey C, Llorca J, Gonzalez-Gay MA. Gelsolin levels are decreased in ankylosing spondylitis patients undergoing anti-tnf-alpha therapy. Clin Exp Rheumatol. 2014;32:218-224
36. Han C, Zhang L, Zhu X, Tang J, Jin X. Plasma gelsolin levels are decreased and correlate with fibrosis in IgA nephropathy. Exp Biol Med. 2013;238:1318-1327
37. Forsman H, Onnheim K, Andreasson E, Christenson K, Karlsson A, Bylund J, Dahlgren C. Reactivation of desensitized formyl peptide receptors by platelet activating factor: A novel receptor cross talk mechanism regulating neutrophil superoxide anion production. PLoS One. 2013;8:e60169
38. Chan W, Lau L, Kwok K, Law W, Ho JCM, Chu K, Poon RTP. A randomized, double-blind, placebo-controlled, ascending-dose trial of the pharmacokinetics and safety of intravenous infusion of recombinant human plasma gelsolin in acutely ill patients with decreased plasma gelsolin levels. American Thoracic Society. 2011;183:A5601
39. Aoqui C, Cuppari L, Kamimura MA, Canziani ME. Increased visceral adiposity is associated with coronary artery calcification in male patients with chronic kidney disease. Eur J Clin Nutr. 2013;67:610-614
40. Kevin Li-Chun H, Schob S, Zeller MW, Pulli B, Ali M, Wang C, Chiou TT, Tsang YM, Lee PS, Stossel TP, Chen JW. Gelsolin decreases actin toxicity and inflammation in murine multiple sclerosis. J Neuroimmunol. 2015;287:36-42
Author contact
![]() Corresponding author: Chien-Te Lee M.D., Ph.D. Division of Nephrology, Department of Internal Medicine, Kaohsiung Chang-Gung Memorial Hospital, Taiwan. 123 Ta-Pei Road, Niao Sung District, Kaohsiung 833, Taiwan. TEL: 886-7-7317123 EXT 8306 FAX: 886-7-7322402 E-mail: chientelcom.
Corresponding author: Chien-Te Lee M.D., Ph.D. Division of Nephrology, Department of Internal Medicine, Kaohsiung Chang-Gung Memorial Hospital, Taiwan. 123 Ta-Pei Road, Niao Sung District, Kaohsiung 833, Taiwan. TEL: 886-7-7317123 EXT 8306 FAX: 886-7-7322402 E-mail: chientelcom.