Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(1):60-67. doi:10.7150/ijms.13264 This issue Cite
Research Paper
Prevalence and Spectrum of TBX5 Mutation in Patients with Lone Atrial Fibrillation
Zhan-Cheng Wang1 ![]() , Wen-Hui Ji2, Chang-Wu Ruan1, Xing-Yuan Liu3, Xing-Biao Qiu4, Fang Yuan4, Ruo-Gu Li4, Ying-Jia Xu4, Xu Liu4, Ru-Tai Huang5, Song Xue5, Yi-Qing Yang4,6,7
, Wen-Hui Ji2, Chang-Wu Ruan1, Xing-Yuan Liu3, Xing-Biao Qiu4, Fang Yuan4, Ruo-Gu Li4, Ying-Jia Xu4, Xu Liu4, Ru-Tai Huang5, Song Xue5, Yi-Qing Yang4,6,7 ![]()
1. Department of Cardiology, Shanghai Eighth People's Hospital, 8 Caobao Road, Shanghai 200235, China;
2. Department of Internal Medicine, Huajing Community Health Service Center of Xu Hui Distric, 180 Jianhua Road, Shanghai 200231, China;
3. Department of Pediatrics, Tongji Hospital, Tongji University School of Medicine, 389 Xincun Road, Shanghai 200065, China;
4. Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China;
5. Department of Cardiovascular Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China;
6. Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China;
7. Department of Central Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China.
Received 2015-7-16; Accepted 2015-11-9; Published 2016-1-23
Abstract
Atrial fibrillation (AF), the most common type of cardiac rhythm disturbance encountered in clinical practice, is associated with substantially increased morbidity and mortality. Aggregating evidence demonstrates that abnormal cardiovascular development is involved in the pathogenesis of AF. A recent study has revealed that the TBX5 gene, which encodes a T-box transcription factor key to cardiovascular development, was associated with AF and atypical Holt-Oram syndrome. However, the prevalence and spectrum of TBX5 mutation in patients with lone AF remain unclear. In this study, the coding regions and splicing junction sites of TBX5 were sequenced in 192 unrelated patients with lone AF and 300 unrelated ethnically-matched healthy individuals used as controls. The causative potential of the identified TBX5 variation was evaluated by MutationTaster and PolyPhen-2. The functional effect of the mutant TBX5 was assayed by using a dual-luciferase reporter assay system. As a result, a novel heterozygous TBX5 mutation, p.H170D, was identified in a patient, with a mutational prevalence of approximately 0.52%. This mutation, which was absent in the 300 control individuals, altered the amino acid completely conserved evolutionarily across species, and was predicted to be disease-causing. Functional deciphers showed that the mutant TBX5 was associated with significantly reduced transcriptional activity when compared with its wild-type counterpart. Furthermore, the mutation significantly decreased the synergistic activation between TBX5 and NKX2-5 or GATA4. The findings expand the mutational spectrum of TBX5 linked to AF and provide new evidence that dysfunctional TBX5 may contribute to lone AF.
Keywords: Atrial fibrillation, Genetics, Transcription factor, TBX5, Reporter gene assay.
Introduction
Atrial fibrillation (AF), the most common type of cardiac arrhythmia encountered in the setting of clinical practice, accounts for approximately one-third of hospitalizations for miscellaneous cardiac rhythm disorders [1]. The estimated prevalence of AF is 1% in the general population, and increases with advancing age, rising from less than 1% in persons under 60 years of age to nearly 10% in people aged over 80 years [1]. AF is responsible for substantially increased morbidity and mortality, conferring a five-fold increased risk of thromboembolic stroke and a two-fold increased risk of demise [2, 3]. Moreover, the incidence of AF-associated stroke also increases with advancing age, rising from 1.5% of subjects in their fifties to 23.5% in octogenarians [1, 3]. Additionally, AF may result in degraded quality of life, poor exercise performance, impaired cognitive function, reduced pulmonary function, tachycardia-induced cardiomyopathy, myocardial infarction, left ventricular dysfunction and congestive heart failure [1,4-7]. Therefore, the social and economic burden of AF is enormous and will continue to increase in the future [8], which warrants continued efforts to identify the etiologies underpinning AF.
AF is frequently associated with various cardiac and systemic conditions, including coronary heart disease, rheumatic heart disease, congenital heart disease, cardiac surgery, pulmonary heart disease, dilated cardiomyopathy, myocarditis, hypertension, chronic renal disease, hyperthyroidism, metabolic disorders, obstructive sleep apnea, non-steroidal anti-inflammatory drug use, and even cancers [1,9-23]. However, in 2% to 45% of AF patients, there are no apparent pathologic substrates for AF, and such AF is referred to as lone AF [1, 24]. A growing body of evidence has convincingly demonstrated that genetic defects play a key role in the pathogenesis of lone AF, and a long list of mutations in over 30 AF-associated genes have been reported, including KCNQ1, KCNH2, KCNE1-5, KCNJ2, KCNA5, SCN5A, KCND3, GJA5, GJA1, ANP, and SCN1-4B [25-43]. Nevertheless, these established AF-associated genes seem to be uncommon causes of AF, and in an overwhelming majority of cases, the genetic determinants underling AF remain unclear.
Aggregating evidence shows that cardiovascular developmental abnormality is a common important anatomic substrate for AF [44]. Studies in developmental biology have revealed the pivotal role of several transcription factors in the normal cardiovascular development, including homeobox transcription factors NKX2-5, NKX2-6 and PITX2, zinc finger transcription factors GATA4, GATA5 and GATA6, and T-box transcription factors TBX1, TBX3, TBX5 and TBX20 [45-48], and a number of mutations in the genes NKX2-5, NKX2-6, PITX2c, GATA4, GATA5 and GATA6 have been causally linked to AF [43,49-59]. Interestingly, recent studies associated a common sequence variation in TBX5 with enhanced susceptibility to AF [60-62], and a TBX5 mutation was identified in patients with AF and atypical Holt-Oram syndrome [63]. However, the prevalence and spectrum of TBX5 mutation in patients with lone AF is still to be evaluated.
Materials and Methods
Ethics
This study was conducted in conformity with the ethical principles of the revised Declaration of Helsinki (Somerset West, Republic of South Africa, 1996). The study protocol was reviewed and approved by the local institutional ethics committee, and written informed consents were obtained from all participants prior to the study.
Study subjects
This study included a cohort of 192 unrelated patients with lone AF and a total of 300 ethnically-matched, unrelated healthy individuals used as controls. They were enrolled from the Chinese Han population. All the study subjects underwent comprehensive physical examination, routine biological test, standard 12-lead electrocardiogram and trans-thoracic echocardiogram. X-ray and coronary angiography were performed only when indicated. The clinical data including medical records, electrocardiogram and echocardiography reports were collected and reviewed. Subjects with hypertension, ischemic heart diseases, congenital heart disease, rheumatic heart disease, diabetes, metabolic diseases, or any other known risk factor of AF were excluded from the current study. The study subjects were clinically classified according to the 2014 AHA/ACC/HRS guideline for the management of patients with AF [1]. Briefly, lone or idiopathic AF was defined as AF occurring in the absence of other cardiac or systemic diseases; familial AF, lone AF occurred in two or more first-degree relatives of a family; paroxysmal AF, AF that terminated spontaneously or with intervention within 7 days of onset; persistent AF, AF lasting more than 7 days; longstanding persistent AF, continuous AF of >12 month duration; permanent AF was used when a joint decision was made by the patient and clinician to cease further attempts to restore and/or maintain sinus rhythm.
Genetic analysis
Peripheral venous blood samples were taken from the study participants and genomic DNA was extracted from white blood cells using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). The referential genomic DNA sequence of TBX5 was from GenBank (GenBank ID: NG_007373.1), a gene sequence database at the National Center for Biotechnical Information (NCBI; http://www.ncbi.nlm.nih.gov/). The intronic primer pairs used to amplify the coding regions and splicing junctions of TBX5 by polymerase chain reaction (PCR) were designed as previously described [64,65]. The TBX5 gene was scanned for potential sequence variation by direct PCR-sequencing in 192 unrelated patients with lone AF and 300 unrelated control individuals. PCR was carried out using HotStar Taq DNA Polymerase (Qiagen, Hilden, Germany) on a Veriti Thermal Cycler (Applied Biosystems, Foster, CA, USA) with standard conditions and concentrations of reagents. The amplified products were purified with the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). The amplicons were sequenced under an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems, Foster, CA, USA) with BigDye® Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems, Foster, CA, USA). The sequencing primers were the same as those used for exonic amplifications. DNA sequences were analyzed with the DNA Sequencing Analysis Software v5.1 (Applied Biosystems, Foster, CA, USA). A sequence variance was verified by bi-directional re-sequencing of an independent PCR-generated amplicon from the same subject. For an identified sequence variance, several databases including the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/), the NCBI′s Single Nucleotide Polymorphism (SNP; http://www.ncbi.nlm.nih.gov/snp) database and PubMed Database (http://www.ncbi.nlm.nih.gov/pubmed) were queried to confirm its novelty.
Multiple alignments of TBX5 protein sequences
To evaluate whether an altered amino acid was evolutionarily conserved, the amino acid sequences of TBX5 in human were aligned with those in chimpanzee, monkey, dog, cattle, mouse, rat, fowl, zebrafish and frog by using the HomoloGene and Show Multiple Alignment links on the NCBI's web site (http://www.ncbi.nlm.nih.gov/homologene).
Prediction of the causative potential of TBX5 sequence variation
The disease-causing potential of a TBX5 sequence variation was predicted by the online programs of MutationTaster (http://www.mutationtaster.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), automatically giving a probability score for each alteration to be either pathogenic or benign.
Expression plasmids and site-directed mutagenesis
The recombinant expression plasmid TBX5-pcDNA3.1, which contains the full-length cDNA of human TBX5, was constructed as described previously [64,65]. The mutant TBX5-pcDNA3.1 was generated by PCR-mediated site-directed mutagenesis using a complimentary pair of primers and QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA), and was validated by sequencing. The recombinant expression plasmids NKX2-5-pEFSA and GATA4-pSSRa, and the atrial natriuretic factor (ANF)-luciferase reporter plasmid, which contains the 2600-bp 5'-flanking region of the ANF gene and expresses the Firefly luciferase, namely ANF-luc, were generous gifts from Dr. Ichiro Shiojima, at the Department of Cardiovascular Science and Medicine, Chiba University Graduate School of Medicine, Chiba, Japan.
Luciferase reporter gene assays
COS-7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% Penicillin/Streptomycin, in an atmosphere with 5% CO2 at 37°C. Transient tranfections were performed in triplicate using the Lipofectamine® 2000 transfecting reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions. To normalize transfection efficiency, the internal control vector pGL4.75 (hRluc/CMV, Promega) expressing the Renilla luciferase was used in transfection assays. COS-7 cells at about 90% confluence were transiently transfected with 0.5 μg of wild-type TBX5-pcDNA3.1, NKX2-5-pEFSA, GATA4-pSSRa, or mutant TBX5-pcDNA3.1, alone or together, in combination with 1.0 μg of ANF-luc and 0.04 μg of pGL4.75. Cells were harvested and lysed 48 h after transfection, and the Firefly luciferase and Renilla luciferase activities were measured with the lysates and the Dual-Glo luciferase assay system (Promega, Madison, WI, USA). The activity of the ANF promoter was presented as fold activation of Firefly luciferase relative to Renilla luciferase.
Statistics
The SPSS version 17.0 software package (SPSS Inc, Chicago, IL, USA) was used to make statistical analyses. Data are expressed as means ± SD, unless otherwise indicated. Continuous variables were tested for normal distribution and Student's unpaired t test was used for the comparison of numeric variables between two groups. Comparison of the categorical variables between two groups was made using Pearson's χ2 test or Fisher's exact test when appropriate. A two-tailed p value < 0.05 indicated significant differences.
Results
Baseline clinical characteristics of the study population
In this study, 192 unrelated patients with lone AF was clinically evaluated in comparison with 300 ethnically-matched, unrelated healthy individuals. None of them had known traditional risk factors for AF. There were no significant differences between patient and control groups in baseline characteristics including age, gender, ethnicity, body mass index, blood pressure, serum lipid, fasting blood glucose, left atrial dimension and left ventricular ejection fraction. The baseline clinical characteristics of the 192 patients with lone AF are summarized in Table 1.
Baseline clinical characteristics of the 192 patients with lone atrial fibrillation.
| Variables | Statistics |
|---|---|
| Baseline demographics | |
| Age at enrollment for the current study (years) | 56 ± 10 |
| Age at initial diagnosis of atrial fibrillation (years) | 50 ± 12 |
| Male (%) | 107 (56) |
| Body mass index (kg/m2) | 24 ± 3 |
| Systolic blood pressure (mmHg) | 132 ± 9 |
| Diastolic blood pressure (mmHg) | 84 ± 6 |
| Left ventricular ejection fraction (%) | 64 ± 5 |
| Left atrial diameter (mm) | 38 ± 4 |
| Personal history of atrial fibrillation (%) | |
| Classification of atrial fibrillation at clinical presentation | |
| Paroxysmal | 131 (68) |
| Persistent | 30 (16) |
| Longstanding persistent | 19 (10) |
| Permanent | 12 (6) |
| History of cardioversion | 119 (62) |
| Positive family history of atrial fibrillation | 57 (30) |
| Medical history (%) | |
| History of syncope | 23 (12) |
| History of pacemaker | 13 (7) |
| History of stroke or transient ischemic attack | 9 (5) |
| Medications (%) | |
| Amiodarone | 142 (74) |
| Warfarin | 100 (52) |
| Aspirin | 31 (16) |
| Digitalis | 29 (15) |
| Beta-blocker | 19 (10) |
| Calcium channel blocker | 6 (3) |
Data are expressed as means and standard deviations, number, or percentage.
Identification of a novel TBX5 mutation
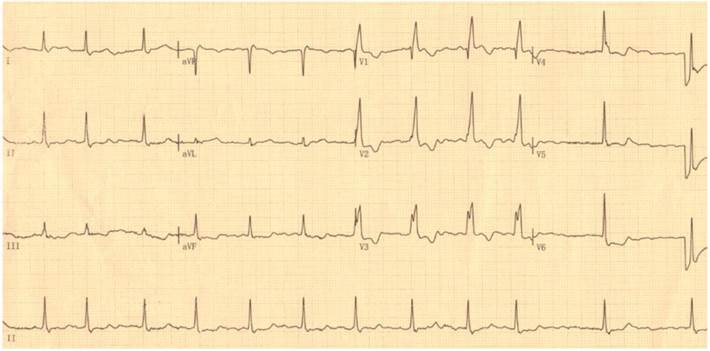
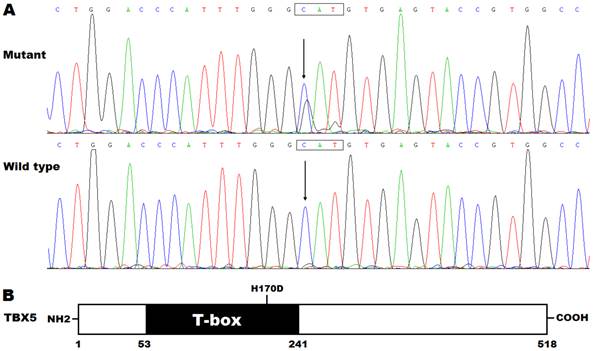
By DNA sequencing, a heterozygous mutation in TBX5 was identified in one of 192 unrelated patients with lone AF, with a mutational prevalence of approximately 0.52%. Specifically, a substitution of guanine (G) for cytosine (C) in the first nucleotide of codon 170 (c.508C>G), predicting the transversion of histidine (H) into aspartic acid (D) at amino acid position 170 (p.H170D), was detected in a male patient with negative family history. The mutation carrier had no apparent congenital abnormalities in the heart, hands and forearms, and his representative electrocardiogram showing AF was shown in Figure 1. The sequence chromatograms showing the heterozygous TBX5 mutation of c.508C>G and its control sequence are shown in Figure 2A. A schematic diagram of TBX5 showing the structural domains and the location of the detected mutation is illustrated in Figure 2B. The identified TBX5 mutation c.508C>G was absent in the 300 control individuals, and was not found in the HGMD, SNP and PubMed databases (accessed again on July 16, 2015), indicating that it is a novel mutation.
Standard 12-lead surface electrocardiogram recorded from the mutation carrier. The representative electrocardiogram shows atrial fibrillation as well as complete right bundle branch block.
Novel TBX5 mutation associated with atrial fibrillation. (A) Sequence electropherograms showing the TBX5 mutation in contrast to its wild type. The arrow indicates the heterozygous nucleotides of C/G in the mutation carrier (mutant) or the homozygous nucleotides of C/C in the corresponding control individual (wild type). The rectangle marks the nucleotides constituting a codon of TBX5. (B) Schematic diagram of TBX5 protein structures with the identified mutation marked. The mutation identified in a patient with lone atrial fibrillation is shown above the structural domains. NH2 denotes amino-terminus, and COOH, carboxyl-terminus.
Multiple alignments of TBX5 protein sequences
As shown in Figure 3, alignment of multiple TBX5 proteins across species displayed that the altered histidine at amino acid position 170 was completely conserved evolutionarily, indicating its functional importance.
Alignment of multiple TBX5 protein sequences across various species. The altered histidine at amino acid 170 is completely conserved evolutionarily among various species.

Causative potential of TBX5 sequence variation
The TBX5 mutation was predicted to be disease-causing by MutationTaster, with a p value of approximately 1.000, and this amino acid substitution was also predicted to be possibly damaging by PolyPhen-2, with a score of 0.993 (sensitivity: 0.47; specificity: 0.96), suggesting that mutated TBX5 might contribute to the development of AF in a subset of patients. No SNPs in the altered region were reported in the MutationTaster database.
Decreased transcriptional activity of the mutant TBX5
As shown in Figure 4, biological assays revealed that the same amount of wild-type and H170D-mutant TBX5 activated the ANF promoter by ∼10-fold and ∼3-fold, respectively (wild type vs mutant: t = 7.95435, P = 0.00135), indicating that the H170D-mutant TBX5 has a significantly decreased transcriptional activity compared with its wild-type counterpart.
Functional impairments resulted from TBX5 mutation. Activation of atrial natriuretic factor promoter driven luciferase reporter in COS-7 cells by wild-type TBX5 or H170D-mutant TBX5 showed significantly decreased transcriptional activity by the mutant protein. In the presence of NKX2-5 or GATA4, the induced synergistic activation by H170D-mutant TBX5 was significantly reduced compared with that by wild-type TBX5. Experiments were performed in triplicate, and mean and standard deviations are given. * represents p < 0.005 when compared with its wild-type counterpart.
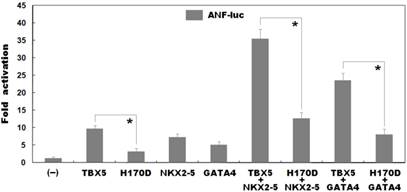
Reduced synergistic transactivation between mutant TBX5 and NKX2-5 or GATA4
As shown in Figure 4, TBX5 in combination with NKX2-5 or GATA4 activated the ANF promoter in a synergistic manner. In the presence of wild-type NKX2-5, the same amount of wild-type and H170D-mutant TBX5 induced activation of the ANF promoter by ∼35-fold and ∼13-fold, respectively (wild type vs mutant: t = 12.2622, P = 0.00025); while in the presence of wild-type GATA4, the same amount of wild-type and H170D-mutant TBX5 induced activation of the ANF promoter by ∼23-fold and ∼8-fold, respectively (wild type vs mutant: t = 9.81299, P = 0.00060). These results suggest that the TBX5 mutation is associated with significantly reduced transactivational activity in synergy with NKX2-5 or GATA4.
Discussion
In the present study, a novel heterozygous TBX5 mutation, p.H170D, was identified in a patient with lone AF. The missense mutation, which was absent in the 600 reference chromosomes, altered the amino acid that was completely conserved evolutionarily across species, and was predicted to be pathogenic by both MutationTaster and PolyPhen-2. Functional deciphers revealed that H170D-mutant TBX5 was associated with a significantly diminished transcriptional activity alone or in synergy with NKX2-5 or GATA4. Therefore, it is probable that the identified TBX5 mutation predisposes the carrier to AF.
The T-box genes encode a family of transcription factors that are characterized by a highly conserved DNA-binding region called T-box. This T-box recognizes and binds specific DNA elements in the promoters of target genes such as ANF and CX40, regulating transcriptional activation or repression. Moreover, the T-box is also a conserved interaction domain for other transcriptionally cooperative partners including NKX2-5 and GATA4 [48]. To date, in the mammalian heart, at least 6 members (TBX1, TBX2, TBX3, TBX5, TBX18 and TBX20) of the T-box gene family have been identified as crucial mediators of myocardial proliferation and patterning [48]. As an important member of the T-box gene family, TBX5 is mapped on human chromosome 12q24.1, coding for a protein with 518 amino acids, which regulates a wide variety of developmental processes in vertebrates and invertebrates, including specification of the mesoderm and development of the heart, vasculature and limbs [48,66]. In the current study, the TBX5 mutation identified in an AF patient is located in the T-box and functional assays unveiled that the mutant protein was associated with substantially reduced transactivation of a target gene, ANF, alone or in combination with NKX2-5 or GATA4. These results suggest that TBX5 haploinsufficiency is likely an alternative pathological mechanism of AF in a minority of patients.
The findings that TBX5 loss-of-function mutation contributes to AF may be attributable at least in part to the abnormal development of the heart. In humans and vertebrates, TBX5 is abundantly expressed in the heart during embryogenesis, playing a pivotal role in cardiovascular development, including myocardial cell proliferation, specification, differentiation, migration, tissue patterning and morphogenesis [48,66-69]. In mice, Tbx5 is widely expressed in cardiac crescent, linear heart tube, common atrium, ventricles, inferior and superior vena cavae, and conduction system, including atrioventricular node and ventricular bundle branches [48,66]. Homozygous disruption of Tbx5 in mice caused embryonic death due to failure of cardiac looping, hypoplasia of sinuatria and left ventricle; while analyses of heterozygous Tbx5-knockout mice demonstrated atrial septal defects, ventricular septal defects, endocardial cushion defects, left heart hypoplasia, and morphological and functional abnormalities in the conduction system, including atrioventricular and bundle branch conduction blocks [69-71]. In embryonic and adult hearts of humans, TBX5 is expressed in the epicardium, myocardium of all four cardiac chambers and endocardium of left ventricle [68], and a number of heterozygous or homozygous TBX5-deficient mutations have been involved in Holt-Oram syndrome, including congenital cardiovascular malformations and cardiac conduction system defects [72]. Furthermore, in humans multiple longitudinal studies have shown that anomalous cardiac conduction system is an independent risk factor for AF [63]. Collectively, these results support that genetically defective TBX5 confers an enhanced susceptibility to AF in humans.
It has been validated that TBX5 transactivates multiple downstream genes, such as ANF and CX40, alone or in synergy with transcriptionally cooperative partners, including NKX2-5 and GATA4 [48,64,73-75], and loss-of-function mutations in some target genes and cooperative partners, including ANF, CX40, NKX2-5 and GATA4, have been associated with AF [43]. Therefore, functionally compromised TBX5 may increase vulnerability to AF by reducing the expressions of some target genes.
It was interesting that both loss-of-function and gain-of-function mutations in TBX5 have been reported to be responsible for Holt-Oram syndrome, with AF being an uncommon clinical manifestation [72,73,76]. Additionally, previous studies have demonstrated that cellular biological sequelae of decreased and increased TBX5 expressions are similar [72,76]. Taken together, these prior findings plus the current data indicate that TBX5 dosage must be finely controlled to avoid cardiovascular diseases [76].
In conclusion, the present study firstly associates TBX5 loss-of-function mutation with lone AF, which expands the mutational spectrum of TBX5 linked to AF and suggests the potential clinical implications for individually tailored treatment of this common arrhythmia.
Acknowledgements
We are really thankful to the participants for their dedication to the study. This work was supported in part by grants from the National Natural Science Fund of China (81470372, 81400244, 81370301 and 81270161), the Natural Science Fund of Shanghai, China (13ZR1438400, 14ZR1438000 and 15ZR1438100), the Key Program for Basic Research of Shanghai, China (14JC1405500), the Experimental animal project of Shanghai, China (14140903600 and 12140902800), and the key project of Shanghai Chest Hospital, China (2014YZDH10102 and 2014YZDH20500).
Conflict of interest
The authors state that there is no conflict of interest in this study.
References
1. January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr, Conti JB, Ellinor PT, Ezekowitz MD, Field ME, Murray KT, Sacco RL, Stevenson WG, Tchou PJ, Tracy CM, Yancy CW; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:e1-76
2. Benjamin EJ, Wolf PA, D'Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946-52
3. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983-8
4. Alegret JM, Viñolas X, Arias MA, Martínez-Rubio A, Rebollo P, Ràfols C, Martínez-Sande JL. New oral anticoagulants vs vitamin K antagonists: benefits for health-related quality of life in patients with atrial fibrillation. Int J Med Sci. 2014;11:680-4
5. Shibata Y, Watanabe T, Osaka D, Abe S, Inoue S, Tokairin Y, Igarashi A, Yamauchi K, Kimura T, Kishi H, Aida Y, Nunomiya K, Nemoto T, Sato M, Konta T, Kawata S, Kato T, Kayama T, Kubota I. Impairment of pulmonary function is an independent risk factor for atrial fibrillation: the Takahata study. Int J Med Sci. 2011;8:514-22
6. Naji F, Pagliaruzzi M, Penko M, Kanic V, Vokac D. Changes in left ventricular filling in patients with persistent atrial fibrillation. Int J Med Sci. 2013;10:1876-9
7. Chu CY, Lin TH, Hsu PC, Lee WH, Lee HH, Chiu CA, Su HM, Lee CS, Yen HW, Voon WC, Lai WT, Sheu SH. Heart rate significantly influences the relationship between atrial fibrillation and arterial stiffness. Int J Med Sci. 2013;10:1295-300
8. Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH Jr, Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, Murray CJ. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014;129:837-47
9. Bengtson LG, Chen LY, Chamberlain AM, Michos ED, Whitsel EA, Lutsey PL, Duval S, Rosamond WD, Alonso A. Temporal trends in the occurrence and outcomes of atrial fibrillation in patients with acute myocardial infarction (from the Atherosclerosis Risk in Communities Surveillance Study). Am J Cardiol. 2014;114:692-7
10. Zhou X, Du JL, Yuan J, Chen YQ. Statins therapy can reduce the risk of atrial fibrillation in patients with acute coronary syndrome: a meta-analysis. Int J Med Sci. 2013;10:198-205
11. O'Neal WT, Efird JT, Dawood FZ, Yeboah J, Alonso A, Heckbert SR, Soliman EZ. Coronary artery calcium and risk of atrial fibrillation (from the multi-ethnic study of atherosclerosis). Am J Cardiol. 2014;114:1707-12
12. Tsai WC, Chen CY, Kuo HF, Wu MT, Tang WH, Chu CS, Lin TH, Su HM, Hsu PC, Jhuo SJ, Lin MY, Lee KT, Sheu SH, Lai WT. Areca nut chewing and risk of atrial fibrillation in Taiwanese men: a nationwide ecological study. Int J Med Sci. 2013;10:804-11
13. Roetker NS, Chen LY, Heckbert SR, Nazarian S, Soliman EZ, Bluemke DA, Lima JA, Alonso A. Relation of systolic, diastolic, and pulse pressures and aortic distensibility with atrial fibrillation (from the Multi-Ethnic Study of Atherosclerosis). Am J Cardiol. 2014;114:587-92
14. Qureshi W, Soliman EZ, Solomon SD, Alonso A, Arking DE, Shah A, Gupta DK, Wagenknecht LE, Herrington D. Risk factors for atrial fibrillation in patients with normal versus dilated left atrium (from the Atherosclerosis Risk in Communities Study). Am J Cardiol. 2014;114:1368-72
15. Xu D, Murakoshi N, Sairenchi T, Irie F, Igarashi M, Nogami A, Tomizawa T, Yamaguchi I, Yamagishi K, Iso H, Ota H, Aonuma K. Anemia and reduced kidney function as risk factors for new onset of atrial fibrillation (from the Ibaraki prefectural health study). Am J Cardiol. 2015;115:328-33
16. Fu XX, Zhao N, Dong Q, Du LL, Chen XJ, Wu QF, Cheng X, Du YM, Liao YH. Interleukin-17A contributes to the development of post-operative atrial fibrillation by regulating inflammation and fibrosis in rats with sterile pericarditis. Int J Mol Med. 2015;36:83-92
17. Mazurek T, Kiliszek M, Kobylecka M, Skubisz-Głuchowska J, Kochman J, Filipiak K, Królicki L, Opolski G. Relation of proinflammatory activity of epicardial adipose tissue to the occurrence of atrial fibrillation. Am J Cardiol. 2014;113:1505-8
18. Zhang Y, Wang YT, Shan ZL, Guo HY, Guan Y, Yuan HT. Role of inflammation in the initiation and maintenance of atrial fibrillation and the protective effect of atorvastatin in a goat model of aseptic pericarditis. Mol Med Rep. 2015;11:2615-23
19. Sharma A, Einstein AJ, Vallakati A, Arbab-Zadeh A, Walker MD, Mukherjee D, Homel P, Borer JS, Lichstein E. Risk of atrial fibrillation with use of oral and intravenous bisphosphonates. Am J Cardiol. 2014;113:1815-21
20. Liu G, Yan YP, Zheng XX, Xu YL, Lu J, Hui RT, Huang XH. Meta-analysis of nonsteroidal anti-inflammatory drug use and risk of atrial fibrillation. Am J Cardiol. 2014;114:1523-9
21. Tamariz L, Hernandez F, Bush A, Palacio A, Hare JM. Association between serum uric acid and atrial fibrillation: a systematic review and meta-analysis. Heart Rhythm. 2014;11:1102-8
22. Yaranov DM, Smyrlis A, Usatii N, Butler A, Petrini JR, Mendez J, Warshofsky MK. Effect of obstructive sleep apnea on frequency of stroke in patients with atrial fibrillation. Am J Cardiol. 2015;115:461-5
23. O'Neal WT, Lakoski SG, Qureshi W, Judd SE, Howard G, Howard VJ, Cushman M, Soliman EZ. Relation between cancer and atrial fibrillation (from the REasons for Geographic and Racial Differences in Stroke Study). Am J Cardiol. 2015;115:1090-4
24. Jahangir A, Lee V, Friedman PA, Trusty JM, Hodge DO, Kopecky SL, Packer DL, Hammill SC, Shen WK, Gersh BJ. Long-term progression and outcomes with aging in patients with lone atrial fibrillation: a 30-year follow-up study. Circulation. 2007;115:3050-6
25. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251-4
26. Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005;16:394-6
27. Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, Jensen HK, Haunsø S, Svendsen JH, Schmitt N. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med Genet. 2012;13:24
28. Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R, Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J, Chen Y. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75:899-905
29. Lundby A, Ravn LS, Svendsen JH, Hauns S, Olesen SP, Schmitt N. KCNE3 mutation V17M identified in a patient with lone atrial fibrillation. Cell Physiol Biochem. 2008;21:47-54
30. Ma KJ, Li N, Teng SY, Zhang YH, Sun Q, Gu DF, Pu JL. Modulation of KCNQ1 current by atrial fibrillation-associated KCNE4 (145E/D) gene polymorphism. Chin Med J (Engl). 2007;120:150-4
31. Ravn LS, Aizawa Y, Pollevick GD, Hofman-Bang J, Cordeiro JM, Dixen U, Jensen G, Wu Y, Burashnikov E, Haunso S, Guerchicoff A, Hu D, Svendsen JH, Christiansen M, Antzelevitch C. Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm. 2008;5:427-35
32. Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings P, Barhanin J, Chen Y. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332:1012-9
33. Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185-91
34. Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, George AL Jr, Roden DM. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927-35
35. Olesen MS, Refsgaard L, Holst AG, Larsen AP, Grubb S, Haunsø S, Svendsen JH, Olesen SP, Schmitt N, Calloe K. A novel KCND3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc Res. 2013;98:488-95
36. Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677-88
37. Thibodeau IL, Xu J, Li Q, Liu G, Lam K, Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, Nicholson BJ, Gollob MH. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation. 2010;122:236-44
38. Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett JC Jr, Olson TM. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158-65
39. Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ, Roden DM. Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2009;2:268-75
40. Wang P, Yang Q, Wu X, Yang Y, Shi L, Wang C, Wu G, Xia Y, Yang B, Zhang R, Xu C, Cheng X, Li S, Zhao Y, Fu F, Liao Y, Fang F, Chen Q, Tu X, Wang QK. Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem Biophys Res Commun. 2010;398:98-104
41. Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu X, Fang WY, Yang YQ. Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation. Int J Mol Med. 2013;32:144-50
42. Olesen MS, Andreasen L, Jabbari J, Refsgaard L, Haunsø S, Olesen SP, Nielsen JB, Schmitt N, Svendsen JH. Very early-onset lone atrial fibrillation patients have a high prevalence of rare variants in genes previously associated with atrial fibrillation. Heart Rhythm. 2014;11:246-51
43. Hong K, Xiong Q. Genetic basis of atrial fibrillation. Curr Opin Cardiol. 2014;29:220-6
44. Mommersteeg MT, Christoffels VM, Anderson RH, Moorman AF. Atrial fibrillation: a developmental point of view. Heart Rhythm. 2009;6:1818-24
45. Akazawa H, Komuro I. Cardiac transcription factor Csx/Nkx2-5: its role in cardiac development and diseases. Pharmacol Ther. 2005;107:252-68
46. Franco D, Christoffels VM, Campione M. Homeobox transcription factor Pitx2: The rise of an asymmetry gene in cardiogenesis and arrhythmogenesis. Trends Cardiovasc Med. 2014;24:23-31
47. Pikkarainen S, Tokola H, Kerkelä R, Ruskoaho H. GATA transcription factors in the developing and adult heart. Cardiovasc Res. 2004;63:196-207
48. Greulich F, Rudat C, Kispert A. Mechanisms of T-box gene function in the developing heart. Cardiovasc Res. 2011;91:212-22
49. Huang RT, Xue S, Xu YJ, Zhou M, Yang YQ. A novel NKX2.5 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. 2013;31:1119-26
50. Xie WH, Chang C, Xu YJ, Li RG, Qu XK, Fang WY, Liu X, Yang YQ. Prevalence and spectrum of Nkx2.5 mutations associated with idiopathic atrial fibrillation. Clinics (Sao Paulo). 2013;68:777-84
51. Yu H, Xu JH, Song HM, Zhao L, Xu WJ, Wang J, Li RG, Xu L, Jiang WF, Qiu XB, Jiang JQ, Qu XK, Liu X, Fang WY, Jiang JF, Yang YQ. Mutational spectrum of the NKX2-5 gene in patients with lone atrial fibrillation. Int J Med Sci. 2014;11:554-63
52. Wang J, Zhang DF, Sun YM, Li RG, Qiu XB, Qu XK, Liu X, Fang WY, Yang YQ. NKX2-6 mutation predisposes to familial atrial fibrillation. Int J Mol Med. 2014;34:1581-90
53. Yang YQ, Xu YJ, Li RG, Qu XK, Fang WY, Liu X. Prevalence and spectrum of PITX2c mutations associated with familial atrial fibrillation. Int J Cardiol. 2013;168:2873-6
54. Zhou YM, Zheng PX, Yang YQ, Ge ZM, Kang WQ. A novel PITX2c loss-of-function mutation underlies lone atrial fibrillation. Int J Mol Med. 2013;32:827-34
55. Wang J, Zhang DF, Sun YM, Yang YQ. A novel PITX2c loss-of-function mutation associated with familial atrial fibrillation. Eur J Med Genet. 2014;57:25-31
56. Qiu XB, Xu YJ, Li RG, Xu L, Liu X, Fang WY, Yang YQ, Qu XK. PITX2C loss-of-function mutations responsible for idiopathic atrial fibrillation. Clinics (Sao Paulo). 2014;69:15-22
57. Wang J, Sun YM, Yang YQ. Mutation spectrum of the GATA4 gene in patients with idiopathic atrial fibrillation. Mol Biol Rep. 2012;39:8127-35
58. Wang XH, Huang CX, Wang Q, Li RG, Xu YJ, Liu X, Fang WY, Yang YQ. A novel GATA5 loss-of-function mutation underlies lone atrial fibrillation. Int J Mol Med. 2013;31:43-50
59. Li J, Liu WD, Yang ZL, Yang YQ. Novel GATA6 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. 2012;30:783-90
60. Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA, Jonasdottir A, Mathiesen EB, Njølstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Løchen ML, Kong A, Thorsteinsdottir U, Stefansson K. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117-22
61. Zang X, Zhang S, Xia Y, Li S, Fu F, Li X, Wang F, Zhang R, Tian X, Gao L, Zhang J, Yang Y, Tu X, Wang Q. SNP rs3825214 in TBX5 is associated with lone atrial fibrillation in Chinese Han population. PLoS One. 2013;8:e64966
62. Sinner MF, Tucker NR, Lunetta KL, Ozaki K, Smith JG, Trompet S, Bis JC, Lin H, Chung MK, Nielsen JB, Lubitz SA, Krijthe BP, Magnani JW, Ye J, Gollob MH, Tsunoda T, Müller-Nurasyid M, Lichtner P, Peters A, Dolmatova E, Kubo M, Smith JD, Psaty BM, Smith NL, Jukema JW, Chasman DI, Albert CM, Ebana Y, Furukawa T, Macfarlane PW, Harris TB, Darbar D, Dörr M, Holst AG, Svendsen JH, Hofman A, Uitterlinden AG, Gudnason V, Isobe M, Malik R, Dichgans M, Rosand J, Van Wagoner DR; METASTROKE Consortium; AFGen Consortium, Benjamin EJ, Milan DJ, Melander O, Heckbert SR, Ford I, Liu Y, Barnard J, Olesen MS, Stricker BH, Tanaka T, Kääb S, Ellinor PT. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation. 2014;130:1225-35
63. Postma AV, van de Meerakker JB, Mathijssen IB, Barnett P, Christoffels VM, Ilgun A, Lam J, Wilde AA, Lekanne Deprez RH, Moorman AF. A gain-of-function TBX5 mutation is associated with atypical Holt-Oram syndrome and paroxysmal atrial fibrillation. Circ Res. 2008;102:1433-42
64. Zhang XL, Qiu XB, Yuan F, Wang J, Zhao CM, Li RG, Xu L, Xu YJ, Shi HY, Hou XM, Qu XK, Xu YW, Yang YQ. TBX5 loss-of-function mutation contributes to familial dilated cardiomyopathy. Biochem Biophys Res Commun. 2015;459:166-71
65. Zhou W, Zhao L, Jiang JQ, Jiang WF, Yang YQ, Qiu XB. A novel TBX5 loss-of-function mutation associated with sporadic dilated cardiomyopathy. Int J Mol Med. 2015;36:282-8
66. Stennard FA, Harvey RP. T-box transcription factors and their roles in regulatory hierarchies in the developing heart. Development. 2005;132:4897-910
67. Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, Bonnet D, Lyonnet S, Young ID, Raeburn JA, Buckler AJ, Law DJ, Brook JD. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet. 1997;15:21-9
68. Hatcher CJ, Goldstein MM, Mah CS, Delia CS, Basson CT. Identification and localization of TBX5 transcription factor during human cardiac morphogenesis. Dev Dyn. 2000;219:90-5
69. Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, Seidman CE. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev Biol. 1999;211:100-8
70. Moskowitz IP, Pizard A, Patel VV, Bruneau BG, Kim JB, Kupershmidt S, Roden D, Berul CI, Seidman CE, Seidman JG. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development. 2004;131:4107-16
71. Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709-21
72. Al-Qattan MM, Abou Al-Shaar H. Molecular basis of the clinical features of Holt-Oram syndrome resulting from missense and extended protein mutations of the TBX5 gene as well as TBX5 intragenic duplications. Gene. 2015;560:129-36
73. Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443-7
74. Linhares VL, Almeida NA, Menezes DC, Elliott DA, Lai D, Beyer EC, Campos de Carvalho AC, Costa MW. Transcriptional regulation of the murine Connexin40 promoter by cardiac factors Nkx2-5, GATA4 and Tbx5. Cardiovasc Res. 2004;64:402-11
75. Zhang Y, Rath N, Hannenhalli S, Wang Z, Cappola T, Kimura S, Atochina-Vasserman E, Lu MM, Beers MF, Morrisey EE. GATA and Nkx factors synergistically regulate tissue-specific gene expression and development in vivo. Development. 2007;134:189-98
76. McDermott DA, Hatcher CJ, Basson CT. Atrial fibrillation and other clinical manifestations of altered TBX5 dosage in typical Holt-Oram syndrome. Circ Res. 2008;103:e96
Author contact
![]() Corresponding authors: Zhan-Cheng Wang, Department of Cardiology, Shanghai Eighth People's Hospital, 8 Caobao Road, Shanghai 200235, China. Tel: +86-21-34284588, fax: +86-21-64514326, E-mail: wang_zhanchengcom; Yi-Qing Yang, Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. Tel: +86-21-62821990, fax: +86-21-62821105, E-mail: yang99yang66com.
Corresponding authors: Zhan-Cheng Wang, Department of Cardiology, Shanghai Eighth People's Hospital, 8 Caobao Road, Shanghai 200235, China. Tel: +86-21-34284588, fax: +86-21-64514326, E-mail: wang_zhanchengcom; Yi-Qing Yang, Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. Tel: +86-21-62821990, fax: +86-21-62821105, E-mail: yang99yang66com.