Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(9):737-741. doi:10.7150/ijms.12517 This issue Cite
Short Research Communication
Heatstroke Effect on Brain Heme Oxygenase-1 in Rats
Ya-Ting Wen1, Tsung-Ta Liu2, Yuh-Feng Lin3, Chun-Chi Chen4, Woon-Man Kung5,6, Chi-Chang Huang7, Tien-Jen Lin1,8, Yuan-Hung Wang3,9, ![]() , Li Wei1,10
, Li Wei1,10 ![]()
1. Department of Neurosurgery, Taipei Medical University-Wan Fang Hospital, Taipei 11696, Taiwan
2. Department of Biology and Anatomy, National Defense Medical Center, Taipei 114, Taiwan
3. Graduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University, Taipei 11031, Taiwan
4. Division of Nephrology, Tri-Service General Hospital, National Defense Medical Center, Taipei 114, Taiwan
5. Department of Neurosurgery, Lo-Hsu Foundation, Lotung Poh-Ai Hospital, Luodong, Yilan 265, Taiwan
6. Department of Exercise and Health Promotion, College of Education, Chinese Culture University, Taipei 11114, Taiwan
7. Graduate Institute of Sports Science, National Taiwan Sport University, Taoyuan 33301, Taiwan
8. Graduate Institute of Injury Prevention and Control, College of Public Health and Nutrition, Taipei Medical University, Taipei 11031, Taiwan
9. Division of General Surgery, Department of Urology, Shuang Ho Hospital, Taipei Medical University, New Taipei City 23561, Taiwan
10. The PhD Program of Translational Medicine, College of Medicine, Taipei Medical University, Taipei 11031, Taiwan
Received 2015-4-27; Accepted 2015-8-9; Published 2015-9-4
Abstract
Exposure to high environmental temperature leading to increased core body temperature above 40°C and central nervous system abnormalities such as convulsions, delirium, or coma is defined as heat stroke. Studies in humans and animals indicate that the heat shock responses of the host contribute to multiple organ injury and death during heat stroke. Heme oxygenase-1 (HO-1)—a stress-responsive enzyme that catabolizes heme into iron, carbon monoxide, and biliverdin—has an important role in the neuroprotective mechanism against ischemic stroke. Here, we investigated the role of endogenous HO-1 in heat-induced brain damage in rats. RT-PCR results revealed that levels of HO-1 mRNA peaked at 0 h after heat exposure and immunoblot analysis revealed that the maximal protein expression occurred at 1 h post-heat exposure. Subsequently, we detected the HO-1 expression in the cortical brain cells and revealed the neuronal cell morphology. In conclusion, HO-1 is a potent protective molecule against heat-induced brain damage. Manipulation of HO-1 may provide a potential therapeutic approach for heat-related diseases.
Keywords: heat stroke, heme oxygenase-1, cortical brain
Brief Communication
The phenomenon of global warming is becoming increasingly serious [1]. One of the consequences of the global warming is an increase in the frequency and intensity of heat waves. Exposure to extremely hot environment (such as among fire fighters, miners, or during a military training) or hot weather can induce heat-related diseases such as heat cramps, heat exhaustion, and heatstroke (HS). HS is defined clinically as a condition when core body temperature rises above 40ºC (hyperthermia) and is accompanied with the central nervous system abnormalities and multi-organ dysfunction [2]. The relative risk for 1-year mortality of hyperthermic versus normothermic patients was 3.4 (95% confidence interval, 1.6 to 7.3) in a cohort study [3], and survivors may remain with the neurologic injury after discharging from the hospital [4]. During the past decade, research showed that HS results from thermoregulatory failure coupled with altered expression of heat shock proteins [5].
Heme oxygenase-1 (HO-1), also named heat shock protein 32, is an important enzyme that degrades heme, a pro-oxidant, leading to the formation of antioxidant molecules [6]. Enzyme reaction of HO-1 generates biliverdin, carbon monoxide (CO), and divalent iron as products, each of which is biologically active and ameliorates the oxidative susceptibility of cells through distinct mechanisms [7]. Previous studies indicated that pretreatment with HO-1 induction material or its gene transfection markedly protects cells and tissues against oxidative damages in vitro and in vivo [8-9]. Although many studies showed the evidence of protective role of HO-1 in their investigations [9], a recent study revealed another data that suggest HO-1 aggravates heat stress-induced neuronal injury in cerebellar Purkinje cells [10]. Hence, this study aims to investigate whether HS induces HO-1 and the location of HO-1 expression in the brain.
Induction of HO-1 mRNA in brain tissue after HS
As shown in Figure 1, we investigated HO-1 expression levels of mRNA in the rat brain after HS. Heat exposure caused a significant induction of HO-1 after HS0. Image analysis of ethidium bromide stained gels indicated that the level of HO-1 mRNA began to increase in HS0 rats and returned to normal in HS6 rats. In addition, quantitative results of RT-PCR (n = 5 in each group) exhibited a similar time course of HO-1 expression. RT-PCR results revealed that levels of HO-1 mRNA peaked at 0 h (2.81 ± 1.25-folds, compared with sham rats) after heat exposure and returned to basal level at 6 h (0.59 ± 0.6-folds, compared with sham rats).
HO-1 mRNA expression in brain tissue at different time frame after heat stroke (HS). Upper panel, image of an ethidium bromide-stained gel showing the time course of HO-1 mRNA expression in brain tissue from rats sacrificed at various recovery time intervals (HS0, HS1, HS3, and HS6). β-actin was used as an internal control for mRNA loading. Lower panel, real-time PCR results of HO-1 mRNA levels at different time points after HS induction. HO-1 mRNA levels were significantly increased in HS0 and HS1 rats (n = 5, * p < 0.05, compared with sham rats).
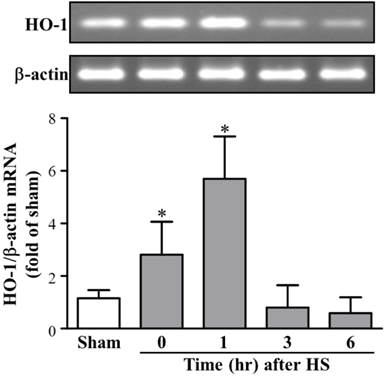
A previous study has shown that the mRNA levels of HO-1 and heat shock protein 70 showed different expression time profiles with a tissue-specific distribution in brain tissues in a gram negative bacterial lipopolysaccharide-induced sepsis [11]; it suggested that the gene expression of these heat shock proteins is separately regulated. The up-regulation of these cytoprotective genes in a specific tissue such as that in the kidney could prevent tissue damage during the period of brain death, and it could be the part of a protective or recuperative mechanism induced by brain death-associated stress [12]. In a rat vasospasm model, researchers reported that the HO-1 mRNA expression was prominently induced in the basilar artery and other brain tissues. They also demonstrated that HO-1 gene induction in cerebral vasospasm after subarachnoid hemorrhage protects against cerebral vasospasm [13].
Protein expression of HO-1 in brain tissue after HS
We further examined the HO-1 protein expression levels in rat brains after HS. As shown in Figure 2, the western blot data (n = 5 in each group) also showed a significant increase in protein levels of HO-1 in HS1 rats (5.30 ± 0.30-folds, as compared with sham rats) and sustained levels in HS3 rats (4.43 ± 0.41-folds, compared with sham rats). Therefore, the time course of steady state levels for HO-1 protein in animals that underwent HS showed that a sustained increase in HO-1 protein was expressed during the observation period of up to 6 hours.
Protein expression of HO-1 in brain tissue at different time frame after heat stroke (HS). Upper panel, representative immunoblot showing the time course of relative HO-1 expression in brain tissue from sham and HS rats (HS0, HS1, HS3, and HS6). β-actin was used as an internal control for protein loading. Lower panel, quantification of protein band densities of HO-1 showed significant increases in HS1, HS3, and HS6 rats (n = 5, * p < 0.05, compared with sham rats).
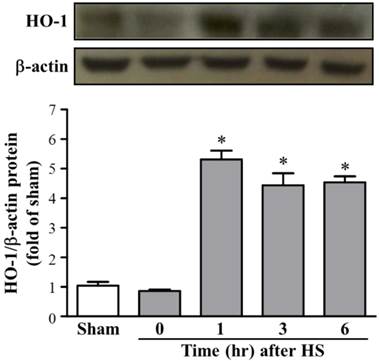
In the present study, the HO-1 mRNA levels increased at 0 h after HS challenge and continued to rise until 1 h, returning to the control level by 3 h after the HS. The HO-1 protein expression level was significantly increased during 1 to 6 h after HS challenge. The time course of HS-induced steady state mRNA and protein levels of HO-1 in the brain are rational and consistent with previous study that has demonstrated the expression pattern of HO-1 in hemorrhagic hypotension and resuscitation-exposed rat livers [14]. HO-1 metabolizes heme to CO, ferrous ion, and biliverdin. CO could promote vasodilation by activating cGMP, and bilirubin, derived from biliverdin, could also serve as an anti-oxidant, both of which may contribute to the reported protective role of HO-1 in cerebral ischemia and subarachnoid hemorrhage [15].
Observation of HO-1 expression in cortical brain after HS
To further characterize the brain cell types expressing HO-1 protein that underwent HS, samples of cortical brain sections from sham or HS rats were stained immunohistochemcally to localize the induction of HO-1. As shown in Figure 3, representative micrograph showed minimal induction of HO-1 in sham rats and continued to increase in HS1 rats. Cells expressing HO-1 revealed neuronal-like cell morphology.
Immunohistochemical staining of HO-1 in brain tissue from sham and heat stroke (HS) rats. Nuclei were stained with hematoxylin (blue), and HO-1 was stained with mouse monoclonal antibody (brown). Left: Low power image (4×) showing location of observed cells. Many cells expressed HO-1 in cortical tissue after HS. Right: high power images (40×) demonstrating that HO-1 was induced in neuronal-like cells following HS.
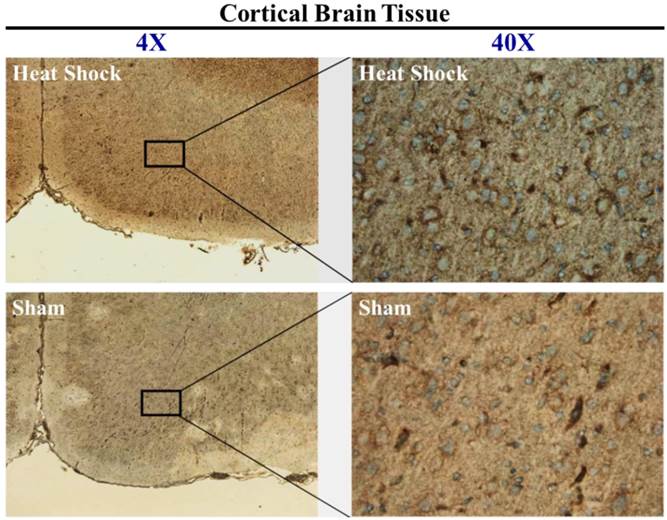
In conclusion, we found a remarkable level of HO-1 mRNA and protein upregulation and its time course in the brain tissue, which may have been closely correlated with the occurrence of delayed brain damage after the heatstroke. As neuroprotective strategy, the therapeutic gene modulation of HO-1 may represent a novel approach for the prevention and treatment of such HS-induced pathologic conditions.
Materials and Methods
Animals and experimental design
Adult male Sprague-Dawley rats (10-12 weeks old) were purchased from the Animal Resource Center of the National Science Council in the Republic of China (Taipei, Taiwan) and housed in the National Defense Medical Center's Center for Laboratory Animal Care. Animals were allowed ad libitum access to food and water, and they were kept in a 12-h day-night cycle. All experimental protocols were approved by the Animal Use and Care Committee of the National Defense Medical Center (Taipei, Taiwan) in accordance with the NIH guidelines for the care and use of laboratory animals.
We have previously established an animal model of HS [16]. In brief, rats were anesthetized by a single intraperitoneal dose of urethane and pentobarbital (0.6 g/kg and 30 mg/kg, respectively). A thermocouple probe was inserted into the rectum for continuous monitoring of the core temperature (Tco), the right femoral artery was cannulated with polyethylene tubing (PE 50) for blood pressure monitoring, and a pressure transducer was inserted into the cannula for continuous monitoring of both mean arterial pressure and heart rate. Rats were subsequently exposed to an ambient temperature of 40°C in a heating chamber until Tco reached 40.5°C (HS onset). After heat exposure, rats were transferred from the heating chamber to an animal cage (room temperature: 25°C), where they remained for various periods of time, permitting the recovery of the core temperature to normal. The recovery time is determined as the number of hours after HS onset: HS3 refers to a condition 3 h after HS onset. HS and normothermic sham rats were sacrificed after various recovery periods with an overdose of urethane.
Real-time reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cortical brain using the TRIzol® reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). Total RNA (3 μg) was reverse transcribed to cDNA using the Rever Tra Ace-a First-strand cDNA Synthesis Kit (Toyobo Life Sciences, Osaka, Japan). The resulting cDNA was incubated with the SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and primers for HO-1 or β-actin (each at 150 nM final concentration).
PCR primer list
| Gene name | Forward | Reverse | Product size (bp) |
|---|---|---|---|
| HO-1 | 5'-CAGAAGGGTCAGGTGTCCAG-3' | 5'-GAAGGCCATGTCCTGCTCTA-3' | 262 |
| β-actin | 5'-CCCATCTATGAGGGTTACGC-3' | 5'-TTTAATGTCACGCACGATTTC-3' | 150 |
For quantitative analysis, we performed 40 amplification cycles (denaturation: 95ºC, 15 s; annealing: 60°C, 30 s; elongation: 72°C, 35 s) on an ABI 7500 PCR Detection System (Applied Biosystems, USA). Melting curve and sequencing data were used to confirm the specificity of the PCR products. HO-1 mRNA levels were normalized to those of β-actin and subsequently expressed as values relative to the control using the comparative threshold cycle method.
Immunoblotting
Cortical brains of the sham and HS rats were collected. Tissues were lysed in a buffer containing 150 mM NaCl, 20 mM Tris-HCl (pH 7.4), 1% sodium deoxycholate, 0.1% SDS, 1% NP-40, and 10% complete protease inhibitor solution (Roche Molecular Biochemicals, Mannheim, Germany). Twenty micrograms of protein lysate were run on 4%-12% NuPAGE Bis-Tris polyacrylamide gels (Invitrogen). Proteins were transferred from the gels to PVDF membranes (Invitrogen, USA) by electroblotting. Each membrane was incubated in a blocking buffer containing phosphate-buffered saline (PBS; 120 mM NaCl, 2.7 mM KCl, and 0.01 M PB), 3% BSA (Sigma, St. Louis, MO, USA), and 0.04% Tween 20 for at least 1 h. Each membrane was then blocked, incubated with an anti-HO-1 antibody (1:2000; Stressgen Biotechnologies, Victoria, British Columbia, Canada) at 4°C overnight, washed in PBS containing 0.04% Tween 20 (three times, 10 min each) to remove unbound primary antibodies, incubated with an HRP-conjugated secondary antibody (1:10,000 diluted in blocking buffer, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 2 h, and washed in PBS (three times, 10 min each). Labeled proteins were visualized by chemiluminescence (ECL Plus kit, Amersham, Arlington Heights, IL, USA).
Immunohistochemical staining of brain tissue
Brain slicing was preformed after completion of the experiment. The obtained brain specimens were excised and fixed in 4% paraformaldehyde. Paraffin-embedded brain tissues were cut at a thickness of 2-5 μm. Parts of these tissues were stained with hematoxylin and eosin for histological examination, and other parts were used for immunohistochemical staining (NovoLink polymer detection system; Leica Biosystems Newcastle Ltd, UK). A semiquantitative study of immunohistochemical staining was performed using image software (Image scope Ver. 10.2.2.2319; Aperio Technologies, Inc., Vista, CA, USA).
Statistical analysis
The plotted data are means ± standard error of the means. Statistical significance was assessed by one-way analysis of variance followed by a Tukey's post hoc test using the SigmaStat software (Jandel Scientific, San Rafael, CA, USA). A value of p < 0.05 was considered statistically significant.
Acknowledgements
This work was supported by grants from the National Science Council (NSC101-2314-B-038 -058) and from Taipei Medical University - Wanfang Hospital (100TMU-WFH-07), Taiwan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tollefson J. Global-warming limit of 2 °C hangs in the balance. Nature. 2015;520:14-15
2. Leon LR, Bouchama A. Heat stroke. Compr Physiol. 2015;5:611-647
3. Wang Y, Lim LL, Levi C, Heller RF, Fisher J. Influence of admission body temperature on stroke mortality. Stroke. 2000;31:404-409
4. Grogan H, Hopkins PM. Heat stroke: implications for critical care and anaesthesia. Br J Anaesth. 2002;88:700-707
5. Biedenkapp JC, Leon LR. Increased cytokine and chemokine gene expression in the CNS of mice during heat stroke recovery. Am J Physiol Regul Integr Comp Physiol. 2013;305:978-986
6. Akagi R, Takahashi T, Sassa S. Fundamental role of heme oxygenase in the protection against ischemic acute renal failure. Jpn J Pharmacol. 2002;88:127-132
7. Gomes AS, Gadelha GG, Lima SJ, Garcia JA, Medeiros JV, Havt A, Lima AA, Ribeiro RA, Brito GA, Cunha FQ, Souza MH. Gastroprotective effect of heme-oxygenase 1/biliverdin/CO pathway in ethanol-induced gastric damage in mice. Eur J Pharmacol. 2010;642:140-145
8. Gu J, Chi M, Sun X, Wang G, Li M, Liu L, Li X. Propofol-induced protection of SH-SY5Y cells against hydrogen peroxide is associated with the HO-1 via the ERK pathway. Int J Med Sci. 2013;10:599-606
9. Schipper HM, Song W. A heme oxygenase-1 transducer model of degenerative and developmental brain disorders. Int J Mol Sci. 2015;16:5400-5419
10. Li CW, Lin YF, Liu TT, Wang JY. Heme oxygenase-1 aggravates heat stress-induced neuronal injury and decreases autophagy in cerebellar Purkinje cells of rats. Exp Biol Med. 2013;238:744-754
11. Fujiwara T, Takahashi T, Suzuki T, Yamasaki A, Hirakawa M, Akagi R. Differential induction of brain heme oxygenase-1 and heat shock protein 70 mRNA in sepsis. Res Commun Mol Pathol Pharmacol. 1999;105:55-66
12. van Dullemen LF, Bos EM, Schuurs TA, Kampinga HH, Ploeg RJ, van Goor H, Leuvenink HG. Brain death induces renal expression of heme oxygenase-1 and heat shock protein 70. J Transl Med. 2013;11:22
13. Suzuki H, Kanamaru K, Tsunoda H, Inada H, Kuroki M, Sun H, Waga S, Tanaka T. Heme oxygenase-1 gene induction as an intrinsic regulation against delayed cerebral vasospasm in rats. J Clin Invest. 1999;104:59-66
14. Bauer I, Wanner GA, Rensing H, Alte C, Miescher EA, Wolf B, Pannen BH, Clemens MG, Bauer M. Expression pattern of heme oxygenase isoenzymes 1 and 2 in normal and stress-exposed rat liver. Hepatology. 1998;27:829-838
15. Sharp FR, Zhan X, Liu DZ. Heat shock proteins in the brain: role of Hsp70, Hsp 27, and HO-1 (Hsp32) and their therapeutic potential. Transl Stroke Res. 2013;4:685-692
16. Liu TT, Hu CH, Tsai CD, Li CW, Lin YF, Wang JY. Heat stroke induces autophagy as a protection mechanism against neurodegeneration in the brain. Shock. 2010;34:643-648
Author contact
![]() Corresponding authors: Dr. Li Wei: Department of Neurosurgery, Taipei Medical University-Wan Fang Hospital, Taipei, Taiwan. Tel.: +886-2-29307930 (ext. 6942). E-Mail: weiliedu.tw. Dr. Yuan-Hung Wang: Graduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan. Tel.: +886-2-22490088 (ext. 8891). E-Mail: wangcecrcom.
Corresponding authors: Dr. Li Wei: Department of Neurosurgery, Taipei Medical University-Wan Fang Hospital, Taipei, Taiwan. Tel.: +886-2-29307930 (ext. 6942). E-Mail: weiliedu.tw. Dr. Yuan-Hung Wang: Graduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan. Tel.: +886-2-22490088 (ext. 8891). E-Mail: wangcecrcom.