International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907
- Current Issue
- Volume 23; 2026
- Volume 22; 2025
- Volume 21; 2024
- Volume 20; 2023
- Volume 19; 2022
- Archive
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(9):708-718. doi:10.7150/ijms.12032 This issue Cite
Research Paper
Secondhand Smoke Exposure Reduced the Compensatory Effects of IGF-I Growth Signaling in the Aging Rat Hearts
Jia-Ping Wu1, Dennis Jine-Yuan Hsieh2, Wei-Wen Kuo3, Chien-Kuo Han4, Peiying Pai5, Yu-Lan Yeh6,7, Chien-Chung Lin8, V. Vijaya Padma9, Cecilia Hsuan Day10, Chih-Yang Huang1,11,12 ![]()
1. Graduate Institute of Basic Medical Science, China Medical University, Taichung, Taiwan
2. School of Medical Laboratory and Biotechnology, Chung Shan Medical University, Taichung, Taiwan
3. Department of Biological Science and Technology, China Medical University, Taichung
4. Department of Health and Nutrition Biotechnology, Asia University, Taichung, Taiwan
5. Division of Cardiology, China Medical University Hospital, Taichung, Taiwan
6. Department of pathology, Changhua Christian Hospital, Changhua
7. Department of Medical Technology, Jen-Teh Junior College of Medicine, Nursing and Management, Miaoli, Taiwan
8. Orthopaedic Department, Armed Forces General Hospital, Taichung, Taiwan
9. Department of Biotechnology, Bharathiar University, Coimbatore-641 046, India
10. Department of Nursing, Mei Ho University, 23 Pingguang Road, Pingtung 91202, Taiwan
11. School of Chinese Medicine, China Medical University, 91 Hsueh-Shih Road, Taichung 40402, Taiwan
12. Department of Health and Nutrition Biotechnology, Asia University, 500 Lioufeng Road, Taichung 41354, Taiwan
Received 2015-3-4; Accepted 2015-6-9; Published 2015-8-28
Citation:
Wu JP, Hsieh DJY, Kuo WW, Han CK, Pai P, Yeh YL, Lin CC, Padma VV, Day CH, Huang CY. Secondhand Smoke Exposure Reduced the Compensatory Effects of IGF-I Growth Signaling in the Aging Rat Hearts. Int J Med Sci 2015; 12(9):708-718. doi:10.7150/ijms.12032. https://www.medsci.org/v12p0708.htm
Other stylesAbstract
Background: Secondhand smoke (SHS) exposure is associated with increased risk of cardiovascular disease. Aging is a physiological process that involves progressive impairment of normal heart functions due to increased vulnerability to damage. This study examines secondhand smoke exposure in aging rats to determine the age-related death-survival balance.
Methods: Rats were placed into a SHS exposure chamber and exposed to smog. Old age male Sprague-Dawley rats were exposed to 10 cigarettes for 30 min, day and night, continuing for one week. After 4 weeks the rats underwent morphological and functional studies. Left ventricular sections were stained with hematoxylin-eosin for histopathological examination. TUNEL detected apoptosis cells and protein expression related death and survival pathway were analyzed using western blot.
Results: Death receptor-dependent apoptosis upregulation pathways and the mitochondria apoptosis proteins were apparent in young SHS exposure and old age rats. These biological markers were enhanced in aging SHS-exposed rats. The survival pathway was found to exhibit compensation only in young SHS-exposed rats, but not in the aging rats. Further decrease in the activity of this pathway was observed in aging SHS-exposed rats. TUNEL apoptotic positive cells were increased in young SHS-exposed rats, and in aging rats with or without SHS-exposure.
Conclusions: Aging reduces IGF-I compensated signaling with accelerated cardiac apoptotic effects from second-hand smoke.
Keywords: Secondhand smoke exposure, aging, age-related death-survival balance, cell cycle, apoptosis.
Introduction
Secondhand smoke (SHS) exposure increases heart disease risk including progressive atherosclerosis, decreased heart rate variability, increased arterial stiffness and increased risk for coronary disease events. Left ventricular hypertrophy has been observed in rabbits exposed to SHS, leading to ventricular remodeling and increased risk for cardiovascular events and mortality [1-3]. SHS is harmful and causes human diseases, especially in children and elderly individuals. Secondhand smoke (SHS) exposure increases the risk for coronary heart disease, especially in elderly individuals, and is associated with increased risk for atherosclerotic heart disease [4]. Old age is a strong independent predictor of death and morbidity in patients with structural heart disease. Therefore, old age is a major risk factor with poor cardiovascular outcome and reduced endogenous cardioprotection [5]. Both the incidence and the severity of atherosclerosis and cardiovascular disease increases with age. The changes to the heart throughout human lifetime are the result of maturational changes beyond sexual maturity, causing myocytes hypertrophy and capillary endothelial cell hyperplasia and interstitial fibroblasts [6]. Age-related cardiac disease is associated with numerous molecular and biochemical changes in the heart. These changes affect protein function and cardiac morphology, resulting in alterations in cell death and cell survival signaling. These biochemical changes also affect mitochondrial membrane anti-apoptosis and apoptosis protein expression levels [7, 8]. Human cardiac aging generates a complex phenotype. Experimental evidence in animal models has indicated attenuation in cardioprotective pathways with aging, yet information regarding myocardial dysfunction in old age smoking is limited. No similar data are available regarding age-related changes in the human smoking heart. Some papers have reported SHS exposure is always associated with cardiovascular disease, especially in old age [9]. Age-related changes in old-age are associated with cardiac diseases including myocardial infarction, aortic regurgitation and alterations to cardiac valves and coronary arteries. SHS exposure involves the combination of the smoke emitted by the burning end of a tobacco cigarette and the smoke exhaled by the smoker into the environment [10-12]. SHS exposure is indicated by elevated serum cotinine and nicotine. Left ventricular pathological hypertrophy due to SHS exposure observed in old age leads to left ventricular remodeling and loss of function [13]. Left ventricular hypertrophy (LVH) is an initial adaptive response. There are many compensatory mechanisms that respond to increased cardiac work-load, sustained left ventricular stimulation being one of them [14]. During LVH development unbalanced progressive remodeling occurs at the cellular level, involving cardiomyocyte survival and cell death or cell loss due to mitochondrial damage [15]. This study further describes the molecular mechanisms involved in SHS exposure in the elderly to identify the pathological underpinnings of cardiac disease and disorders.
Apoptosis, or programmed cell death, is a recognized mechanism for the elimination of redundant cells in the pathogenesis of human cardiac disorders in the elderly [16]. Cardiac IGF-I triggers intracellular signaling cascades that are involved in modulating and facilitating growth and survival and promotes apoptosis [17]. The death-receptor-induced apoptotic pathway is initiated by death-agonists and involves the Fas ligand, (Fas-L/Fas-FADD-caspase 8-Bid/t-Bid) reportedly involved in the pathogenesis of LHV. The mitochondria plays an important role in apoptosis by releasing cytochrome c and active caspase 9. However, caspase 3 apoptosis signaling mediates both mitochondria-dependent and death-receptor-dependent apoptotic pathways [18]. In cardiomyocytes, insulin-like growth factor (IGF-I) activates PI3K (phosphatidylinositol-3-kinase)/Akt (PKB, protein kinase B) signaling through IGF-IR and is considered to play a role in preventing myocyte apoptosis [19]. The important role of IGF-I and IGF-IR in growth and development and their involvement in the prevention of cell apoptosis have been elucidated. In cardiomyocytes, PI3k activity is required for IGF-I and its receptor (IGF-IR), and PI3K-generated phospholipids regulate AKT activity by direct binding of phosphoinositides to the PH domain [20].
The present investigation determines whether SHS exposure and aging causes synergistic effects in cardiac cell death pathway activation, including mitochondria-dependent and death-receptor-dependent Fas-signaling, as well as identifying the adaptive IGF-I survival pathway mechanism associated with inflammation circulating markers.
Materials and Methods
Animals
Male Sprague-Dawley rats were purchased from the National Science Council Animal Center, Taipei, Taiwan, young (6 weeks) and old (18 months old). Six animals were housed in individual cages in an environmentally controlled animal room with temperature and humidity controlled chambers (12-h light/12-h dark cycle). Tap water was freely provided with a standard chow diet. All experimental animals were handled in accordance with the Taiwan Society for Laboratory Animals Sciences guide for animal care.
Experimental groups and secondhand smoke (SHS) exposure
After arrival and a brief acclimation period, young and old rats were randomly divided into the following subgroups: control animals not exposed to cigarette smoke, and secondhand smoke (SHS)- animals exposed to 30 min of cigarette SHS twice a day for 4 weeks. Rats were placed in an exposure chamber and exposed to 10 cigarettes.
Histological analysis by Masson's trichrome staining
Left ventricle cross sections were cut at a thickness of 10 µm and placed on slides. Cross sections were stained with Masson's trichrome staining for left ventricle cross-sectional collagen area and extracellular space assessment. Deparaffin embedded slides were soaked in xylene for 10 minutes and then hydrated in 80%, 70%, 65% ethanol for 5 minutes each. Samples were stained with Masson's trichrome to detect left ventricular cross section collagen accumulation. Stained sections were then rinsed with PBS and air dried before mounting. After gently rinsing with water, the slides were rehydrated through a graded alcohol series for 15 min, and then cleaned in xylene.
Western blot and ELISA analysis
Proteins were separated using 10.5~12.5% SDS polyacrylamide gel electrophoresis at 100 V for 1 hr. Electrophoresis proteins were transferred to PVDF membranes using Bio-Rad Mini Trans-Blot Electrophoretic Transfer Cell Instruments at 150 mA for 2 hr. Membranes were blocked in 1~5% non-fat milk in washing buffer (10 mM Tris pH 7.5, 100 mM NaCl, 0.1%Tween-20) for 1 h at room temperature. After washing, cells were probed with Fas-L, Fas, FADD, caspase 8, Bcl 2, Bad, t-Bid, Bid, Cytochrome c, Caspase 9, Caspase 3, IGF-I, IGF-II, p-PI3K, PI3K, p-Akt, Akt, JAK1/2, STAT3, pP38α, P38α, p-JNK1/2, JNK1/2. antibodies (Santa Cruz Lab) for 2 h. After washing, membranes were incubated with enzyme conjugated anti-rabbit or anti-goat, or anti-mouse IgG horseradish peroxidase for 1 h at room temperature in washing buffer. The membranes were then washed in blotting buffer for 10 min three times. Color development was performed using ECL chemiluminescence. Cyclin A, Cyclin B, Cyclin D1 and Cyclin E (both Santa Cruz Lab System) protein concentration in left ventricle tissue supernatants were measured using ELISA according to the manufacturer's protocol.
Terminal Nucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) and DAPI stained
Left ventricular sections from young and old rats were dewaxed by incubating in xylene. Sections were then rehydrated by placing in 95%, 70%, 50%, 30% ethanol for 10 minutes each and preincubated with proteinase k (20ug/ml in 10Mm Tris-HCl pH 7.6) at room temperature. Left ventricular sections were rinsed in TUNEL mixture, at 37 ℃, for one hour in a humidified chamber. After rinsing, sections were stained DAPI. Slides were then mounted with a propidium iodide solution and analyzed under fluorescent microscope.
Statistical analysis
Data were collected and analyzed using SigmaStat software. All data are expressed as the mean±standard error of the mean (SEM). Comparison between groups was conducted using two-way analysis of variance (ANOVA) and following post-hoc using Student-Newman-Keuls analysis. p values less than 0.05 and 0.01 were considered statistically significant and highly statistically significant. *p<0.05, **p<0.01, significant statistic difference compared to young control. #p<0.05, significant statistic difference compared to old control.
Results
Left ventricular remodeling in Old SHS exposure rats
Secondhand smoke (SHS) exposure is associated with various cardiovascular disease (CVD) risk factors. We examined changes in histological and morphological cardiac or the left ventricle using Masson's trichrome to investigate whether SHS exposure resulted in cardiac remodeling. Left ventricular cross sections results showed collagen accumulation and ECM degradation in young SHS exposure, old control, old SHS exposure rat hearts (Figure 1A). Compared with the young control, with aging, both age-related morphological and pathophysiologic cardiovascular changes were predisposed to left ventricular hypertrophy (LVH) or heart failure (HF) (Figure S1). Substantial collagen tissue enlargement existed as a result in young SHS exposure, old and old SHS exposure rat hearts. Left ventricular interstitial muscle fibers were broad and disordered as well (Figure 1A). Left ventricular cross sections from old SHS exposure exhibited more ECM remodeling. Echocardiographic assessment demonstrated left ventricle cardiac remodeling more prevalent than in other parts of the whole heart (Figure S1B and S1C). Collagen accumulation was observed in the blue area. The morphometric samples are shown at x400 magnification using Masson's trichrome staining. Highly structured collagen fibers formed an intricate myocardium. Collagen fibers were easily seen in the old rat hearts. Quantitative and statistical analysis determined the percentage of extracellular space and collagen content in the tissue section area (Figures 1B and 1C). We found that the percentage of collagen content obtained in young SHS exposure, old control and old SHS exposure rat hearts was significantly higher than that in the young control (Figure 1B). We also found the TIMPs content in the old SHS exposure rat hearts was significantly higher than that in the old control (p<0.05 vs. old control) (Figure S2B). In order to determine whether cardiac remodeling occurred in the hypertrophied aging rat hearts, we found changes in the extracellular space percentage significantly increased in young SHS exposure (p<0.05), old (p<0.05) and the old SHS exposure (p<0.01) rat hearts, compared with the young control (Figure 1C). The objective of this work was to study the MMPs content in the left ventricular hypertrophy as a cardiac function of aging (Figure S1 and Figure S2A). The same result was found when compared with aging. We could determine the physiological aging or pathological SHS exposure changes in the heart leading to cardiac remodeling.
Upregulation of death receptor dependent apoptosis pathway (TNFα/Fas-L-Fas/FADD-cleaved caspase 8) occurs in old rats and SHS-exposed animals
The aging heart is accompanied with left ventricular apoptosis involving left ventricular myocytes being constantly replaced by newly formed myocytes from birth to death. This aging process offers changing the balance between cell death and cell growth. Given the above evidence that both SHS exposure and aging lead to left ventricular remodeling and hypertrophy, to further explore the induced death receptor dependent apoptosis pathway mechanism in the young control, young SHS exposure, old control and old SHS exposure rats cell death receptor dependent apoptosis pathway protein expression levels were examined, including Fas, Fas-L, FADD and Caspase 8 by western blot analysis (Figure 2A). Fas-L/Fas is a cell death signal. Fas-L from extracellular apoptosis signal was increased in rats exposed to SHS and the old control rat left ventricle, when compared with the young control. However, Fas-FADD-Caspase 8 is a intercellular apoptosis signal. These signals modulate Fas and FADD activity. Fas was increased in young SHS exposed (p<0.05), old SHS exposed (p<0.01) and old control rats (p<0.05). We found that Fas was more dramatically increased in old rats exposed to SHS than in old control rats (p<0.05). The other protein, FADD, was increased only in rats exposed to SHS, both in young and old animals. In contrast, a comparison of FADD levels in old rats compared to young control rats returned a significant finding. Interestingly, significantly higher levels of caspase 8, a downstream target of Fas/FADD, were found in old control (p<0.05) (Figure 2B). Similarly, cleaved-caspase 8 was increased in SHS exposure groups, including both young (p<0.05) and old controls (p<0.01). We detected the caspase 8-cutting proteins Bid and t-Bid, both of which exhibited increased levels only in old control and old SHS-exposed rats (Figure 3). However, we did not find alterations in Bid and t-Bid protein expression in young SHS exposure groups. Therefore, we could identify a difference between cell death or from mitochromal apoptosis signaling pathway.
Figure 1
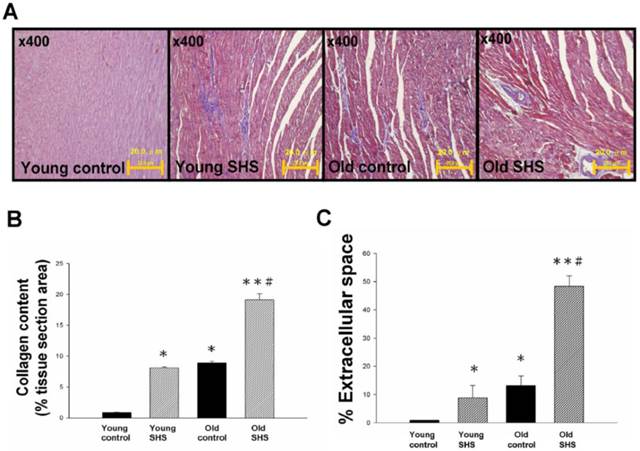
Myocardial extracellular matrix and cardiac remodeling disruption in young SHS exposure, old and the old SHS exposure rat hearts. (A) Representative images and histological analysis of heart total collagen content using Masson's trichrome staining. Morphological features of cardiac remodeling in young SHS exposure, old and old SHS exposure rat hearts. The left ventricular architecture images were magnified 400x. Resolution with 20μm calibration bar. (B) Quantitative analysis of left ventricular cross-sectional area of collagen content. Densitometric graphs represent means ± SEM. *p<0.05, **p<0.01, significant difference compared to young control. #p<0.05, significant difference compared to old control. (C) Quantitative analysis of left ventricular cross-sectional area of extra-cellular space. Bars represent means ± SEM (N=6). *p<0.05, **p<0.01, significant statistic difference compared to young control. #p<0.05, significant statistic difference compared to old control.
Figure 2
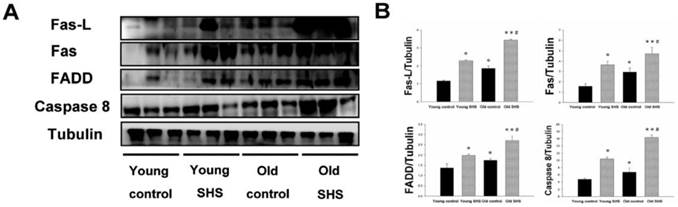
A possible non-mitochondria dependent mechanism for apoptosis with aging or with SHS exposure. (A) Representative western blotting of Fas-L, Fas, FADD and Caspase 8 protein expression levels in young, young SHS exposure, old and old SHS exposure rat left ventricles. (B) All protein expression levels were quantified using densitometry. Tubulin protein levels showed protein loading. Data was quantified in densitometry and expressed as means ± SEM (N=6). *p<0.05, **p<0.01 significant statistic difference compared with young control. #p<0.05, significant statistic difference compared with old control.
Figure 3
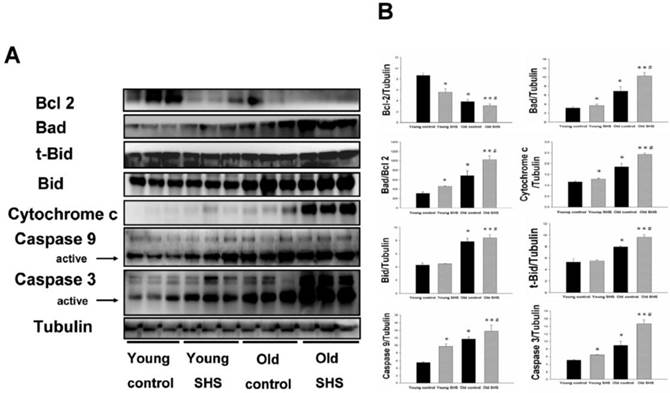
Mitochondrial apoptosis protein expression augmented in old rat hearts exposure to SHS. (A) Representative western blotting analysis of protein expression levels for Bcl 2, Bad, t-Bid, Bid, Cytochrome c, Caspase 9 and Caspase 3 in young control, young SHS exposure, old control and old SHS exposure rats heart. (B) Quantitative analysis of protein densitometry expression levels for Bcl 2, Bad, t-Bid, Bid, Cytochrome c, Caspase 9 and Caspase 3. Results are as the optical density ratio of Bad to Bcl 2, α-Tubulin protein levels showed protein loading. Bars show means ± SEM (N=6). *p<0.05, **p<0.01 significant statistic difference compared with young control. #p<0.05, significant statistic difference compared with old control.
Mitochondria apoptosis proteins were only increased in old rats, and the increase was augmented in old SHS-exposed rats
Mitochondrial damage and dysfunction, which can result in cell death and cell loss, play an important role in aging. Mitochondrial membrane damage due to cytochrome c release from the mitochondria is a key apoptotic event. Particular importance is placed on the role of cytochrome c release from the mitochondria and alterations in apoptotic proteins (such as Bad) and anti-apoptotic proteins (such as Bcl 2). The ratio of Bad/Bcl 2 protein expression was increased in old control rats (p<0.05) and in aging rats exposed to SHS (p<0.01) (Figure 3A) compared to young rats. This result indicated that cytochrome c was being released from the mitochondria during the aging process. Thus, cytochrome c protein expression was increased in old control rats (p<0.05) as well as in aging rats exposed to SHS (p<0.01). However, cytochrome c expression was increased in aging rats exposed to SHS in comparison with old control. (p<0.05) (Figure 3B). In addition, caspase 9 is downstream of the Bcl 2 family apoptotic cascade. Caspase 3 is involved in apoptotic signaling via the Fas-L/Fas and mitochondrial apoptotic cascade (Figures. 2 and 3). Aging increased left ventricular caspase 9 and caspase 3 protein expression. Cleaved caspase 3 and cleaved caspase 9 protein expression were dramatically higher in the left ventricle of aging rats exposed to SHS (p<0.01) compared to the young control group. However, their expression was not altered in the young SHS exposure group.
The cardiac survival pathway IGF-I/IGFIR-PI3K/p-Akt was found to be attenuated in old control and old SHS exposed rats, but not in young rats; especially in rats exposed to SHS
Figure 4
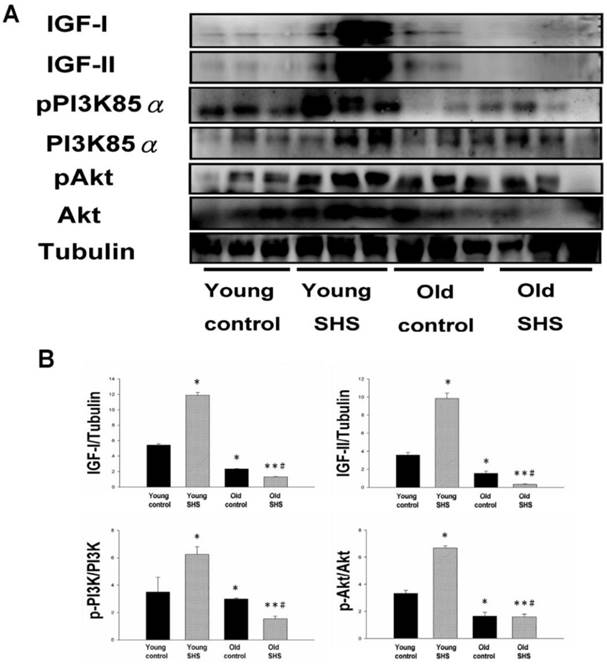
Decline in Insulin-like growth factors, IGF-I and IGF-II, and PI3K/AKT signaling in old and old SHS exposure rats, but elevated in young SHS exposure rat hearts. (A) Representative western blotting analysis of the protein expression levels of IGF-I, IGF-II, pPI3K85α, PI3K85α, pAkt, and Akt in young control, young SHS exposure, old and old SHS exposure rat left ventricles. (B) Quantitative analysis of the protein densitometry expression levels of IGF-I, IGF-II, pPI3K85α, PI3K85α, pAkt, and Akt in young control, young SHS exposure, old control and old SHS exposure rat hearts. Bars show means ± SEM (N=6). *p<0.05, **p<0.01, significant statistic difference compared to young control, #p<0.05, significant statistic difference compared to old control.
IGF-I/IGF-II/PI3K/Akt signaling is one of the best-characterized determinants of longevity in model systems (Figure 4). We observed clear increases in IGF-I and IGF-II in young rats exposed to SHS (p<0.05). Additionally, we observed a decline in the IGF-I and IGF-II in old rats, and even more decreases in old SHS-exposed rats (p<0.01). Both PI3K and AKT and their active form protein expression were also decreased in the old rat heart tissues with or without SHS (Figure 4). On the other hand, the ratio of p-PI3K/PI3K and p-AKT/AKT appeared to decrease in old control and in old rats exposed to SHS (Figure 4B). Moreover, we found that the ratio of p-PI3K/PI3K and p-AKT/AKT were significantly diminished in old rats exposed to SHS compared with young and old control. These finding indicate reduced IGF-I/IGFII-PI3K/p-Akt signaling pathway expression in old rats and cardiac cell proliferation regulation. Overexpression of IGF-I/IGF-II-PI3K/Akt signaling in young SHS exposure rats not only mediated adult left ventricular hypertrophy compared with the young control, but attenuated of IGF-I/IGF-II-PI3K/Akt signaling increased mitochondrial apoptosis protein expression in old SHS exposure rats compared with the old control. These findings suggested that SHS exposure almost completely reconstituted IGF-I/IGF-II-PI3K/Akt dependent cell proliferation in old rats. Furthermore, we checked the number of apoptotic cells in young SHS exposure, old and old SHS exposure by TUNEL staining. We found a dramatic increase in the number of cell groups (Figures 5A and B). This fact led us to think about the possible biochemical molecular events which can regulate the cell death and survival balance. An observation that the magnitude of left ventricular tissue injury was further supported by SHS exposure affecting the survival and cell death signaling pathway balance. To determine whether reduced IGF-I/IGF-II-PI3K/Akt decreased cell proliferation from SHS exposure effects, and also by aging, we detected cell cycle checkpoint protein expression using western blotting. As Figure 6 shows cyclin A, B, D, E protein expression levels were decreased in all treatment groups leading to cell cycle arrest, in contast, cyclin-dependent kinase inhibitor, p21, and its upstream regulator, p53, were by inhibited. We therefore suggest that cell survival and proliferation function is reduced in young SHS exposure, old control and old rats exposed to SHS (Figure 6).
SHS exposure exacerbated circulation in aging inflammatory and age-related diseases
Although systemic insulin resistance and reduced insulin/IGF-I signaling in the heart could alleviate age-associated alterations, we need mounting evidence to determine if SHS exposure induced inflammatory responses that exacerbate cardiac aging and aged-related left ventricular hypertrophy. To examine potential inflammation circulating markers we examined IL-6, TNFα, p38α and Jnk antibodies expression levels using immunochemistry analysis and JAK1/2, STAT3, P38α, pP38α, JNK1/2 and pJNK1/2 using western blotting (Figure 7). These molecules were markedly increased in young SHS exposure, old control and old SHS exposure rats, as disclosed at Western blotting levels. In old rats exposed to SHS, we found positive antibodies expression of JAK1/2, STAT3, P38α, pP38α, JNK1/2 and pJNK1/2. We could suggest SHS exposure exacerbated aging inflammatory and age-related left ventricular hypertrophy.
Figure 5
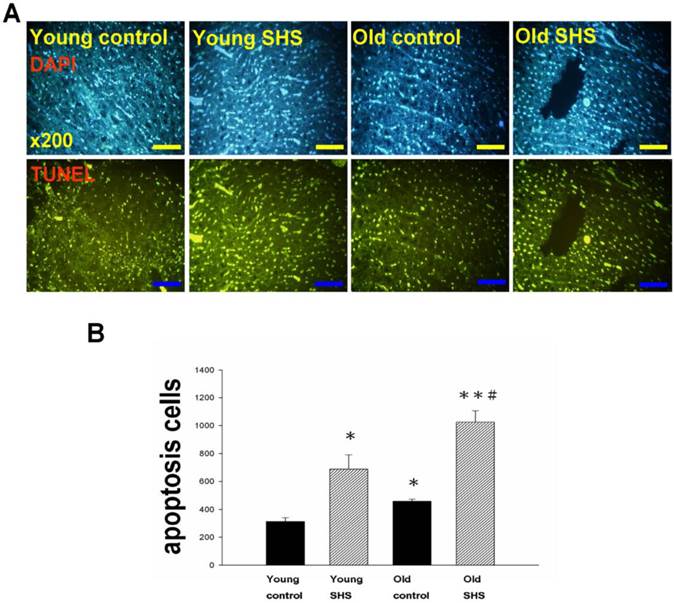
Cell death numbers in young control, young SHS exposure, old control, old SHS exposure rat hearts. (A) DAPI and TUNEL apoptosis analysis. Green express apoptotic cells. (B) Quantitative analysis of apoptotic cells numbers. Bars show means ± SEM (N=6). *p<0.05, **p<0.01, significant statistic difference compared to young control, #p<0.05, significant statistic difference compared to old control.
Figure 6
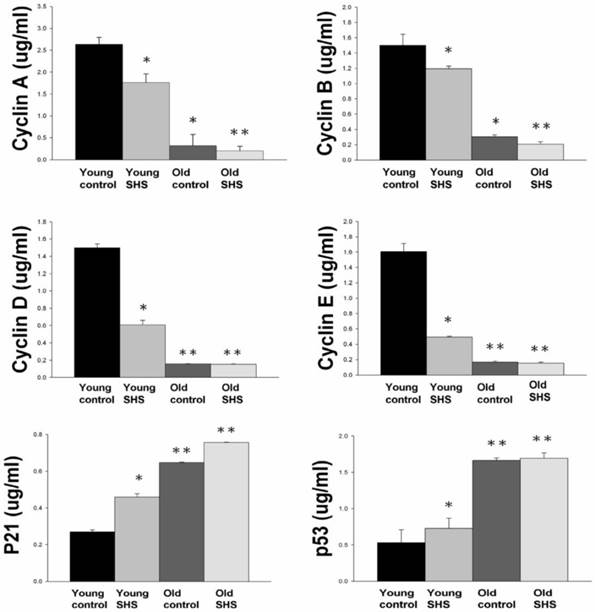
Down regulation of cell cycle resulted in low proliferation and development in old and old exposure to SHS rats. Quantitative analysis of enzyme-linked immunosorbent assay of cyclin A, B, D, E, p21 and p53. Results are the mean ± SEM (N=6). *p<0.05, **p<0.01, significant statistic difference compared to young control. #p<0.05, significant statistic difference compared to old control.
Discussion
Secondhand smoke exposure (SHS) has been linked to a number of harmful health outcomes and is an important cause of morbidity and mortality. There is evidence indicating that SHS exposure presents a challenging health hazard [21]. It is also well understood that toxic air contamination can cause lung cancer and cardiovascular diseases. This study investigated the SHS exposure effects associated with aging, specifically in the left ventricles of male rats. As is well known, old age in humans always accompanies increased incidence of atherosclerotic vascular and cardiovascular disease [22]. However, aging is a physiological process caused by increasing injuries and biological vulnerability that reduces the organisms' ability to survive. Aging affects various aspects of vascular morphology and function and has recently been considered a major risk factor for cardiovascular disease presenting various effects on left vascular morphology and function. Age affects cardiovascular function in the same manner as secondhand smoke exposure. Age-related changes in left ventricular morphology [23] and function include decreased myocyte number, increased myocyte size, increased left ventricular wall thickness, decreased conduction fiber density and increased myocardial stiffness in left ventricular function [24]. Aging also shows relative adaptive responses to eliminate damaged and exhausted cells from birth to senescence [25]. As we know, SHS increases arterial stiffness and coronary cardiac disease events. We found that the changes associated with aging lead to pathological cardiovascular outcomes and the same cardiac adaptations that result from exposure to SHS (Figure 1 and Figure S1). We found similar changes in cardiac morphology in aging rats and rats that underwent SHS exposure. We observed left ventricular fibrosis and increased left ventricular wall thickness (Figure S1 and Figure S2). Left ventricular hypertrophy was found in both aging and SHS exposed rats. At this time point the heart changes associated with age may mimic heart disease, such as cardiac hypertrophy and heart failure. Aging leads to parallel stiffening of the aorta and heart, which causes an increase in systolic stiffness, contractility and diastolic stiffness [26]. Four weeks of SHS exposure to fine particulate matter led to an increase in polymorphonuclear leukocyte cells. In the aging heart left ventricular myocytes are constantly replaced by newly formed myocytes from birth to death. This aging process offers an extraordinary example of the effects that the dynamic balance between cell death and cell growth has on the physiological and pathological restructuring of the heart [27-30]. In the normal heart the rate of cell death increases with age and is not balanced by new myocyte formation. The viable myocytes become hypertrophic to preserve myocardial mass in an aging heart. Thus, myocyte death, hypertrophy, and lack of new myocyte formation characterize the aging heart. Apoptosis is involved in mitochondrial-dependent and non-mitochondrial-dependent pathways. Two apoptosis pathways are consistently observed in both the aging and SHS exposed rat heart.
Apoptosis has been reported to contribute to cardiomyocyte loss that may be involved in cell proliferation control [31, 32]. SHS and aging produce a synergistic activated increase in the Bad/Bcl2 ratio, elevating mitochondrial membrane pore permeability and increased cytochrome c release subsequent to caspase-9 and caspase-3 activation to promote cell death in the heart (Figure 3). Cytokine/Fas receptor-driven pathways for cardiomyocyte apoptosis should also be taken into consideration [33]. In contrast, we found that while aging myocytes are more prone to activating the cell death signaling pathway, younger cells possess hypertrophic ability and are less susceptible to cell death. TNFα/Fas-L and Fas/FADD signaling pathways were consistently increased in the hearts of aging rats and SHS-exposed rats and even more highly activated in the hearts of aging rats exposed to SHS (Figures 2 and 3). Similarly, cleaved-caspase 8 was activated in aging and SHS exposed rat hearts. Activated caspase 8 cleaves Bid to form t-Bid led t-Bid to bind to mitochondrial membranes were found exhibiting increased expression in old rats exposed to SHS.
Figure 7
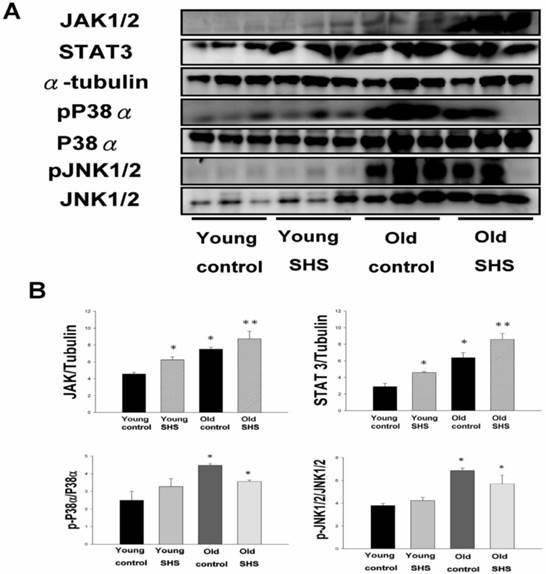
Secondhand smoke (SHS) exposure induced age-related hypetrophy in old rats. (A) Representative western blotting of JAK1/2, STAT3, pP38α, P38α, pJNK1/2, and JNK1/2 protein expression levels in young, young SHS exposure, old and old SHS exposure rat hearts. α-Tubulin protein levels showed protein loading. (B) Quantitative analysis of the protein densitometry expression levels of JAK1/2, STAT3, pP38α, P38α, pJNK1/2, and JNK1/2 protein expression levels. Statistical analysis of the ratio of pP38α to P38α and pJNK1/2 to JNK1/2. Data was quantified in densitometry and be expressed as means ± SEM (N=6). *p<0.05, **p<0.01 significant statistic difference compared to young control. #p<0.05, significant statistic difference compared with old control.
Apoptosis is due to a death-survival imbalance. The PI3K/AKT pathway delivers a survival signal [34], mediated by growth factors such as IGF-I and IGF-II. IGF-I exhibited a potent anti-apoptotic property through the anti-apoptotic member of the Bcl2 family and PI3K-transduced survival signals and their downstream signaling cascade after AKT activation and Bad inactivation [35]. This process occurred only in young SHS-exposed rats and not in aging or aging-plus-SHS-exposure rats (Figure 4). Similar results were observed in our apoptosis positive cells (Figure 5), which revealed an anti-hypertrophic effect in rat hearts. The cell cycle is more easily arrested in older and SHS exposed animals (Figure 6). Overall, we believe SHS and aging both enhance left ventricular hypertrophy (Figure 7). These results indicate that SHS exposure and aging induce mitochondria-dependent and -independent apoptosis signaling pathways upregulation and down regulation of survival signaling pathways.
Conclusions
Taken together these data demonstrate that once aging rats are exposed to SHS the extracellular space in the aging heart is now recognized as an essential element of the myocardial structure and function, and a dynamic participant in remodeling. Aging reduces IGF-I compensated signaling and accelerates the cardiac apoptotic effects induced by second-hand smoke. Aging and SHS should both be considered risk factors for cardiac dysfunction.
Supplementary Material
Figures S1-S2.
Acknowledgements
We thank Dr. Chien-Chung Lin for the use of microscope facility, Chia-Hua Kuo for TUNEL staining, Jia-Ping Wu for technical assistance in imaging studies, and Chih-Yang Huang for critical reading of the manuscript.
Sources of funding
This study was supported in part by the Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH102-TD-B-111-004) and Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of Excellence (MOHW104-TDU-B-212-113002).
Conflict of Interest
All authors have read and approved the manuscript, and the full disclosure of any relationship with industry.
References
1. Barnoya J, Glantz SA. Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation. 2005;111:2684-98
2. Hamer M, Stamatakis E, Kivimaki M. et al. Objectively measured secondhand smoke exposure and risk of cardiovascular disease: what is the mediating role of inflammatory and hemostatic factors? J Am Coll Cardiol. 2010;56:18-23
3. Lu SQ, Fielding R, Hedley AJ. et al. Secondhand smoke (SHS) exposures: workplace exposures, related perceptions of SHS risk, and reactions to smoking in catering workers in smoking and nonsmoking premises. Nicotine Tob Res. 2011;13:344-52
4. Rudatsikira E, Muula AS, Siziya S. Exposure to environmental tobacco smoke among adolescents in Kampala-Uganda. East Afr J Public Health. 2002;6:197-9
5. Weeks SG, Glantz SA, De Marco T. et al. Secondhand smoke exposure and quality of life in patients with heart failure. Arch Intern Med. 2011;171:1887-93
6. Olivetti G, Melissari M, Capasso JM. et al. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68:1560-8
7. Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev. 1993;73:413-67
8. Nadal-Ginard B, Kajstura J, Leri A. et al. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res. 2003;92:139-50
9. Boyle P, Maisonneuve P. Lung cancer and tobacco smoking. Lung Cancer. 1995;12:167-81
10. Venn A, Britton J. Exposure to secondhand smoke and biomarkers of cardiovascular disease risk in never-smoking adults. Circulation. 2007;115:990-5
11. Lakatta EG. Cardiovascular aging research: the next horizons. J Am Geriatr Soc. 1999;47:613-25
12. Raya TE, Gaballa M, Anderson P. et al. Left ventricular function and remodeling after myocardial infarction in aging rats. Am J Physiol. 1997;273:H2652-58
13. Anversa P, Palackal T, Sonnenblick EH. et al. Myocyte cell loss and myocyte cellular hyperplasia in the hypertrophied aging rat heart. Circ Res. 1990;67:871-85
14. Li XM, Ma YT, Yang YN. et al. Down regulation of survival signaling pathways and increased apoptosis in the transition of pressure overload-induced cardiac hypertrophy to heart failure. Clin Exp Pharmacol Physiol. 2009;36:1054-61
15. Jahangir A, Sagar S, Terzic A. Aging and cardioprotection. J Appl Physiol (1985). 2007;103:2120-8
16. Priebe HJ. The aged cardiovascular risk patient. Br J Anaesth. 2000;85:763-78
17. Warner HR. Aging and regulation of apoptosis. Curr Top Cell Regul. 1997;35:107-21
18. Kuo WW, Wu CH, Lee SD. et al. Second-hand smoke-induced cardiac fibrosis is related to the Fas death receptor apoptotic pathway without mitochondria-dependent pathway involvement in rats. Environ Health Perspect. 2005;113:1349-53
19. Phaneuf S, Leeuwenburgh C. Cytochrome c release from mitochondria in the aging heart: a possible mechanism for apoptosis with age. Am J Physiol Regul Integr Comp Physiol. 2002;282:R423-30
20. Wu Y, Wang D, Wang X. et al. Caspase 3 is Activated through Caspase 8 instead of Caspase 9 during H(2)O(2)-induced Apoptosis in HeLa Cells. Cell Physiol Biochem. 2011;27:539-46
21. Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3'-kinase and mitogen-activated protein kinase pathways. J Biol Chem. 1997;272:154-61
22. Centurione L, Di Giulio C, Cacchio M. et al. Correlations between protein kinase C zeta signaling and morphological modifications during rat heart development and aging. Mech Ageing Dev. 2003;124:957-66
23. Wu JP, Che TT. Secondhand Smoke Exposure in Aging-related Cardiac Disease. Aging Dis. 2013;4:127-33
24. Gould NS, Min E, Gauthier S. et al. Aging adversely affects the cigarette smoke-induced glutathione adaptive response in the lung. Am J Respir Crit Care Med. 2010;182:1114-22
25. Wu J, Hsieh C, Ho T. et al. Secondhand smoke exposure toxicity accelerates age-related cardiac disease in old hamsters. BMC Cardiovasc Disord. 2014;14:195
26. Stessman-Lande I, Jacobs JM, Gilon D. et al. Physical activity and cardiac function in the oldest old. Rejuvenation Res. 2012;15:32-40
27. Centurione L, Antonucci A, Miscia S. et al. Age-related death-survival balance in myocardium: an immunohistochemical and biochemical study. Mech Ageing Dev. 2002;123:341-50
28. Sarkar S, Chawla-Sarkar M, Young D. et al. Myocardial cell death and regeneration during progression of cardiac hypertrophy to heart failure. J Biol Chem. 2004;279:52630-42
29. Sun M, Chen M, Dawood F. et al. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:398-407
30. Pollack M, Phaneuf S, Dirks A. et al. The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann N Y Acad Sci. 2002;959:93-107
31. Chisalita SI, Dahlström U, Arnqvist HJ. et al. Increased IGF1 levels in relation to heart failure and cardiovascular mortality in an elderly population: impact of ACE inhibitors. Eur J Endocrinol. 2011;165:891-8
32. Welch S, Plank D, Witt S. et al. Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ Res. 2002;90:41-8
33. Serneri GG, Modesti PA, Boddi M. et al. Cardiac growth factors in human hypertrophy. Relations with myocardial contractility and wall stress. Circ Res. 1999;85:57-67
34. Condorelli G, Drusco A, Stassi G. et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:12333-8
35. Cheng SM, Ho TJ, Yang AL. et al. Exercise training enhances cardiac IGFI-R/PI3K/Akt and Bcl-2 family associated pro-survival pathways in streptozotocin-induced diabetic rats. Int J Cardiol. 2013;167:478-85
Author contact
![]() Corresponding author: Graduate Institute of Basic Medical Science, China Medical University, Taichung, Taiwan, 40402, R.O.C. E-mail: cyhuangcmu.edu.tw; Tel: 886-4-22053366 ext. 3313; Fax:886-4-22051276
Corresponding author: Graduate Institute of Basic Medical Science, China Medical University, Taichung, Taiwan, 40402, R.O.C. E-mail: cyhuangcmu.edu.tw; Tel: 886-4-22053366 ext. 3313; Fax:886-4-22051276
Citation styles
APA
Wu, J.P., Hsieh, D.J.Y., Kuo, W.W., Han, C.K., Pai, P., Yeh, Y.L., Lin, C.C., Padma, V.V., Day, C.H., Huang, C.Y. (2015). Secondhand Smoke Exposure Reduced the Compensatory Effects of IGF-I Growth Signaling in the Aging Rat Hearts. International Journal of Medical Sciences, 12(9), 708-718. https://doi.org/10.7150/ijms.12032.
ACS
Wu, J.P.; Hsieh, D.J.Y.; Kuo, W.W.; Han, C.K.; Pai, P.; Yeh, Y.L.; Lin, C.C.; Padma, V.V.; Day, C.H.; Huang, C.Y. Secondhand Smoke Exposure Reduced the Compensatory Effects of IGF-I Growth Signaling in the Aging Rat Hearts. Int. J. Med. Sci. 2015, 12 (9), 708-718. DOI: 10.7150/ijms.12032.
NLM
Wu JP, Hsieh DJY, Kuo WW, Han CK, Pai P, Yeh YL, Lin CC, Padma VV, Day CH, Huang CY. Secondhand Smoke Exposure Reduced the Compensatory Effects of IGF-I Growth Signaling in the Aging Rat Hearts. Int J Med Sci 2015; 12(9):708-718. doi:10.7150/ijms.12032. https://www.medsci.org/v12p0708.htm
CSE
Wu JP, Hsieh DJY, Kuo WW, Han CK, Pai P, Yeh YL, Lin CC, Padma VV, Day CH, Huang CY. 2015. Secondhand Smoke Exposure Reduced the Compensatory Effects of IGF-I Growth Signaling in the Aging Rat Hearts. Int J Med Sci. 12(9):708-718.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.