ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2015; 12(8):674-679. doi:10.7150/ijms.12450 This issue Cite
Research Paper
Arsenic Trioxide Activate Transcription of Heme Oxygenase-1 by Promoting Nuclear Translocation of NFE2L2
Zhen Yue1#, Lingzhi Zhong1, 3#, Yan Mou1, 2, Xiaotong Wang1, Haiying Zhang1, Yang Wang1, Jianxin Xia2, Ronggui Li1 ![]() , Zonggui Wang2
, Zonggui Wang2 ![]()
1. Key Laboratory of Pathobiology, Ministry of Education, Norman Bethune College of Medicine, Jilin University, Changchun, China
2. The Second Hospital of Jilin University, Changchun, P.R. China.
3. Current address: Institute of Basic Medical Sciences, College of Life Science, Chinese PLA General Hospital, Beijing 100853, China
#These authors contributed equally to this work.
Abstract
In a previous study, we found that induced expression of Heme Oxygenase-1 (HO-1) is responsible for the resistance of human osteosarcoma MG63 cells to the chemotherapeutic agent arsenic trioxide (ATO). The present study was aimed at investigating the molecular mechanisms underlying the induction of HO-1 that occurs after exposure of MG63 cells to ATO. First, using RT-QPCT and Western-blot, we found that ATO strongly induced the expression of heme oxygenase-1 (HO-1) in these human osteosarcoma cells. Then by analyzing HO-1 mRNA of MG63 cells exposed to ATO in the presence and absence of a transcription inhibitor Actinomycin-D (Act-D), we demonstrated that ATO activates HO-1 expression in MG63 cells by regulating the transcription of the gene. Finally, through the analysis of the NFE2L2 protein levels among the total cellular and nuclear proteins by Western-blot and Immunocytochemical staning, we determined that ATO enhanced the nuclear translocation of nuclear factor erythroid 2-like 2 (NFE2L2), also known as Nrf2. From these results we have concluded that transcription activation of HO-1 resulting from the nuclear translocation of NFE2L2 is the underlying molecular mechanism for its high induction, which, in turn, is responsible for the resistance of human osteosarcoma cells to ATO treatment.
Keywords: Arsenic trioxide, heme oxygenase-1, nuclear factor erythroid 2-like 2, osteosarcoma, Nuclear Translocation
Introduction
Osteosarcoma is the most common primary malignant bone tumor in children, adolescents and young adults [1]. Currently accepted treatments for this disease include surgery, chemotherapy, and radiation-therapy [2, 3]. Commonly employed chemotherapeutic agents are Methotrexate (MTX), Adriamycin (ADM), cis-dichlorodiamineplatinum (CDDP), and ifosfamide (IFO) [2]. However, the therapeutic efficiency of these treatments is often transient and ineffective for advanced osteosarcoma [4]. Therefore, there is an urgent requirement to identify new therapies for the disease.
Arsenic trioxide (ATO) is a traditional Chinese medicine which has long been used to treat a wide spectrum of diseases including arthritis, dermatologic conditions, neuropathies, hemorrhoids, and asthma [5]. Recently, ATO has been used to treat various malignancies and has been shown to have significant benefit in the treatment of acute promyelocytic leukemia [6-8] as well as a broad spectrum of hematologic and solid tumors in preclinical models [9, 10]. In the treatment of osteosarcoma, ATO has been shown to induce apoptosis of p53 null human1osteosarcoma MG63 cells through the inhibition of catalase [11] and inhibit proliferation of human osteosarcoma U2OS cells by regulating the gene expression of Survivin and Bcl-2 [12].
Heme oxygenase-1 (HO-1), degrades heme into biliverdin, iron, and CO, is a major antioxidant enzyme [13] and plays a central role in the removal of intracellular reactive oxygen species (ROS) [14]. Because it regulates formation of ROS, a common mediator of apoptosis, the induction of HO-1 could presumably lessen the efficacy of chemotherapeutic agents in the treatment of human osteosarcoma. In this respect, HO-1 has been shown to provide cellular protection in multiple myeloma cells against the key anti-myeloma drugs bortezomib and lenalidomide [15]. In a previous study, we found that induced expression of HO-1 confers resistance of human osteosarcoma MG63 cells to ATO [16]. Little is known about the mechanisms by which ATO induces expression of HO-1 in these cells. Therefore, the present study was carried out to identify the mechanisms underlying the induction of HO-1 that resulted from the exposure of human osteosarcoma MG63 cells to ATO.
Materials and Methods
Materials
ATO was purchased from Pharmaceuticals Limited Company of Harbin Medical University (Harbin, China). High-glucose Dulbecco's Modified Eagle Medium (H-DMEM) was purchased from Gibco BRL (Rockville, USA). Fetal bovine serum (FBS) was purchased from HyClone Inc. (Logan, USA). PCR primers were purchased from Sangon Biotech (Shanghai, China). RNase-free DNase I, Trizol reagent and the RT-reaction kit were purchased from TaKaRa Biotechnology (Dalian, China). Actinomycin D and TSA were from Beyotime Institute of Biotechnology (Jiangsu, China). Rabbit anti-human NFE2L2 polyclonal antibody from Signalway Antibody (Maryland, USA), Mouse anti-human Histone H3 monoclonal antibody from Tianjin Sungene Biotech Co., Ltd. (Tianjin, China), Rabbit anti-human HO-1 polyclonal antibody, mouse anti-human β-actin monoclonal antibody, horseradish peroxidase (HRP)-labeled anti-rabbit IgG and anti-mouse IgG were purchased from Proteintech Group (Chicago, USA). The enhanced chemiluminescence (ECL) kit was purchased from Pierce Biotechnology, Inc. (Rockford, USA).
Cell culture and treatments
Human osteosarcoma MG63 (ATCC#: CRL-1427) and U2OS (ATCC#: HTB-96™) cells were purchased from the American Type Culture Collection (Manassas, USA) Human umbilical vein endothelial cells (HUVECs) from the Central South University Xiangya School of Medicine, (Changsha, China) were maintained in H-DMEM supplemented with 10% heat-inactivated FBS and were incubated in a humidified 5% CO2 incubator at 37°C. For the experiments, the cells were plated in 6 well dishes and incubated for 24 hours. Subsequently the medium was exchanged with fresh medium containing different concentrations of ATO as indicated in respective figures. Vehicle was used to treat the control cells. Cell treatments were conducted for another 24 hours. Actinomycin D (10 μg/ml) was added to the cells 30 min earlier than addition of ATO. TSA (500ng/ml) was added to the cells at the same time as ATO.
RNA purification and RT-QPCR
Total RNA from the treated cells was purified with a TRIzol Reagent following the manufacturer's instruction. The purity and quantity of the RNA were measured with a spectrophotometer and the quality of RNA was monitored by agarose gel electrophoresis. After treatment with RNase-free DNase I, RNA was subjected to reverse transcription with a RT-reaction Kit. The cDNA product was amplified and quantified with 7300 Real-time PCR system (Applied Biosystems) in a 25 μl reaction volume using SYBRR Green PCR Master Mix. The primer sets used for PCR amplification are shown in Table 1. The thermal cycling program consisted of 2 min at 50°C, 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. After amplification, a melting curve was generated and data analysis was performed by Dissociation Curves 1.0 software (Applied Biosystems). The normalized value was expressed as the ratio of mRNA of the target gene to mRNA of the reference gene (β-actin) in each sample. The magnitude of activation was expressed as the ratio of the normalized values of each group treated with ATO to that of the control group.
Protein extraction and western blotting analysis
Total cellular protein was extracted by a modified radioimmunoprecipitation (RIPA) buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 0.5% Na-deoxycholate, 1% NP-40, 1 mM PMSF, 1% Trasylol, 0.1% PEFA and 10% Glycerol). Nuclear protein was extracted by a nuclear protein extraction kit from Signalway Antibody (Maryland, USA), following the manufacturer's instruction. Protein samples were resolved by 10% SDS-PAGE and then were transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, USA). After blocking and washing, the membranes were incubated with primary (rabbit anti-human HO-1 and rabbit anti-human NFE2L2 polyclonal antibodies and mouse anti-human β-actin and mouse anti-human histone H3 monoclonal antibodies, respectively). Following extensive washing, the membranes were incubated with horseradish peroxidase (HRP)-labeled anti-rabbit IgG or HRP-labeled anti-mouse IgG for 1 h. After washing the membranes, the immunoreactive bands were visualized by using an ECL kit, and then the membranes were exposed to film and analyzed by using an automatic gel imaging analysis system (Gene, Cambridge, UK).
Immunocytochemical staining assay
Following the manufacturer's instruction, immunocytochemical staining was carried out by a SP DAB Detection Kit, based on Streptavidin-Peroxidase, purchased from Fuzhou Maixin Biotech Co., Ltd (Fuzhou, China).
Statistical analysis
All calculations and statistical analyses were performed using GraphPad Prism 5.0 software (San Diego, CA, USA). Student's t-test was used to analyze the significance of any differences between two groups. P<0.05 was considered statistically significant.
Results
High induction of HO-1 by ATO in human osteosarcoma cells
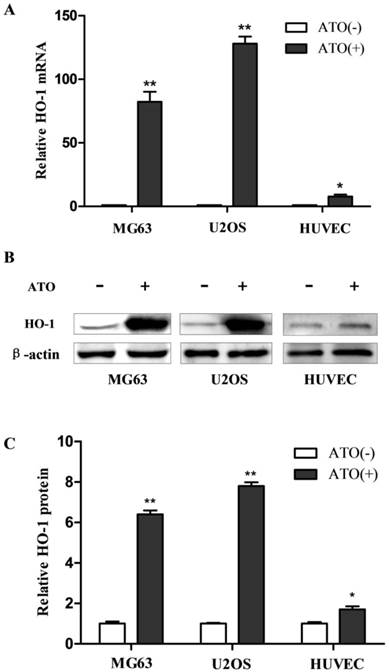
As the first step to investigate the molecular mechanisms underlying the induction of HO-1 from exposure to ATO in the cells, we examined the specificity of the induction. Human osteosarcoma MG63 and U2OS cells and non-malignant human umbilical vein endothelial cells (HUVECs) were exposed to ATO. HO-1 mRNA and protein were analyzed by RT-QPCR and western blot, respectively. As shown in Fig. 1, HO-1 mRNA was increased by more than 80 and 120 fold in human osteosarcoma MG63 and U2OS cells, respectively, but by less than 8 fold in HUVECs (Fig. 1A). HO-1 protein was increased by more than 6 and 8 fold in human osteosarcoma MG63 and U2OS cells, respectively, but increased by less than 2 fold in HUVECs after exposure to ATO (Fig. 1C). These results indicate that high expression of HO-1 induced by the exposure to ATO occurs in human osteosarcoma MG63 and U2OS cells but less marked in HUVECs.
Induction of HO-1 in human osteosarcoma cells following treatment with ATO. Cells were exposed to 10 μM ATO for 24 hours. The mRNA and protein were then analyzed by RT-QPCR and western blot, respectively. The amount of mRNA and protein were normalized to β-actin mRNA or protein. Relative fold activation was calculated based on the ratio of the normalized values of the cells treated with ATO to that of the control cells. The data are expressed as the mean ± SD, N=6, *P<0.05 and **P<0.01 versus control cells without exposure to ATO.
Primer sets used for RT-QPCR
| Genes | Primer sets | Sequenes | GenBank |
|---|---|---|---|
| HO1 | Forward | 5'-CAGTGCCACCAAGTTCAAGC-3' | NM_002133 |
| Reverse | 5'-GTTGAGCAGGAACGCATCTT-3' | ||
| β-actin | Forward | 5'-CATGTACGTTGCTATCCAGGC-3' | NM_001101 |
| Reverse | 5'-CTCCTTAATGTCACGCACGAT-3' |
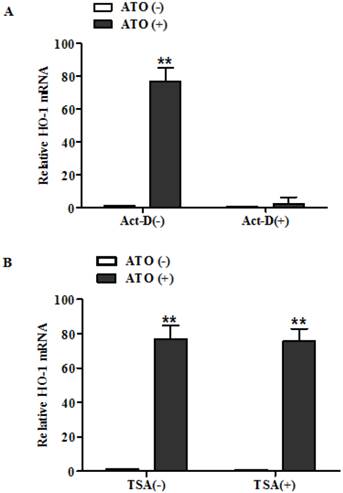
Actinomycin-D abolished HO-1 induction in MG63 cells following their treatment with ATO. Cells were exposed to ATO (10 μM) for 24 hours in the presence of Act-D or vehicle. Act-D (10 μg/ml) was added to the cells 30 min earlier than addition of ATO and TSA (500ng/ml) was added to the cells at the same time as the addition of ATO. HO-1 mRNA was analyzed by RT-QPCR and the amount of mRNA was normalized to β-actin mRNA. Relative fold activation was obtained based on the ratio of the normalized values of the cells treated with ATO to that of the untreated control cells. The data are expressed as the mean ± SD, N=6, **P<0.01 versus control cells without exposure to ATO.
ATO induced HO-1 transcription in MG63 cells
To distinguish between activation of the transcription rate and the suppression of the mRNA turn over as the molecular mechanism underlying the induction of HO-1 after exposure to ATO, we then analyzed HO-1 mRNA of the cells exposed to ATO in the presence and absence of a transcription inhibitor Actinomycin-D (Act-D). We also analyzed HO-1 mRNA in the presence and absence of histone deacetylase inhibitors (TSA) to determine whether histone acetylation is involved in the HO-1 induction from exposure to ATO.
As shown in Fig. 2, HO-1 mRNA was increased by more than 80 fold in human osteosarcoma MG63 (Fig. 2A) in the absence of Act-D, but the high induction was abolished by the transcription inhibitor. These results indicate that the induction of HO-1 from exposure to ATO in MG63 cells resulted from its regulation on the transcription rate of the gene, but not from the suppression of the mRNA turn over. However, histone deacetylase inhibitors TSA had no effects on the induction (Fig. 2B). These results indicate that the histone acetylation is not involved in the HO-1 induction from exposure to ATO.
ATO enhanced the nuclear translocation of NFE2L2
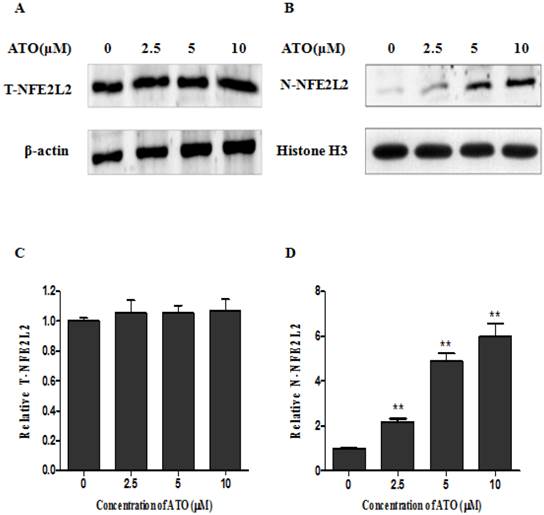
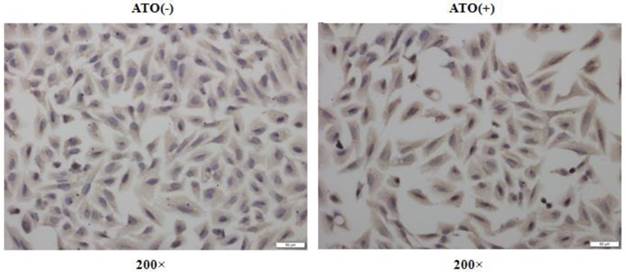
ATO has been frequently reported to induce cellular ROS production in many normal and malignant cells [11, 16, 17]. NFE2L2, as a transcription factor, plays a crucial role in regulating genes which contain antioxidant response elements (ARE) in their promoters [18, 19]; many of these genes encode proteins involved in the removal of intracellular reactive oxygen species (ROS) [18, 19]. To explore the molecular mechanisms underlying the induction of HO-1 after exposure to ATO, we analyzed the expression levels of NFE2L2 in the total cellular and the nuclear proteins by Western-blot and Immunocytochemical staining assay. Fig. 3 shows the changes of NFE2L2 protein levels among the total cellular and nuclear proteins, respectively. A and B are representative photographs of Western-blots. C and D are statistical analysis of the results. Among the total cellular protein there was no significant change in the levels of NFE2L2 (T-NFE2L2 in Fig. 3C) following ATO treatment. However, within the nuclear protein there was a clear dose-dependent increase of NFE2L2 (N-NFE2L2 in Fig. 3D) after the cells were exposed to ATO. These results indicate that the translocation from the cytoplasm to the nucleus of NFE2L2 is an underlying molecular mechanism for the high induction of HO-1 that is responsible for the resistance of human osteosarcoma cells to ATO treatment. The conclusion was further supported by Immunocytochemical staining assay. As shown in Fig. 4, the nuclei of MG63 cells exposed to ATO express stronger NFE2L2 than untreated cells, as visualized by Immunocytochemical staining.
Discussion
The resistance of osteosarcoma cells to chemotherapeutic drugs remains a major factor that limits current treatment modalities for osteosarcoma. In a previous study, we found that induced expression of Heme Oxygenase-1 (HO-1) is responsible for the resistance of human osteosarcoma cells to chemotherapeutic agent ATO [16]. In the present study, we found that ATO induced high expression of HO-1 in human osteosarcoma cells and that the induction was due to the transcriptional regulation of the gene, but not due to changes in the mRNA turn over. HO-1 is a key redox enzyme and plays an important role in the removal of intracellular ROS, which prevents ROS-mediated apoptosis. The transcriptional regulation of HO-1 determines the level of the encoded protein and therefore influences the ability of the cells to remove the intracellular ROS, which further prevents ROS-mediated apoptosis. Our results provide new evidence for a mechanism through which osteosarcoma cells can develop resistance to ATO by enhancing the removal of intracellular ROS and protecting the cells from apoptosis via the induction of HO-1 transcription. These results are consistent with previous reports in which ROS was involved in arsenic induced cell death in human leukemia cells [20], hepatocytes [21] and in testicular tissue of the experimental rats [22].
The exposure to ATO caused nuclear translocation of NFE2L2 in MG63 cells. Cells were exposed to ATO for 24 hours at the concentration indicated in the figure. Total cellular and nuclear proteins were separated by PAGE and subjected to Western blot analysis. For photography and densitometric analysis, Quantity One software (Version 4.6.3, Bio Rad, USA) was used. NFE2L2 proteins in total cellular and nuclear proteins were normalized for loading to β-actin and Histone H3 protein, respectively and expressed relative to the value of the untreated control cells. Fig. 3A and Fig. 3B are representative photographs of Western blots of total cellular and nuclear protein, respectively. Fig. 3C and Fig. 3D are statistical analysis of quantified western blots. The data are expressed as the mean ± SD, N=6, **P<0.01 versus the control cells without exposure to ATO.
NFE2L2 nuclear translocation in MG63 cells induced by the exposure to ATO. Cells were exposed to ATO (10 μM) or vehicle for 24 hours. The expression of NFE2L2 protein was visualized by immunocytochemical staining. The figures are representative photographs of immunocytochemical preparations of the cells with (ATO+) and without (ATO-) exposure.
Gene transcription is regulated through the interactions between the trans-factors and cis-elements in the regulatory region of the gene [23]. NFE2L2 is a transcription factor which is a member of a small family of basic leucine zipper (bZIP) proteins and regulates genes which contain antioxidant response elements (ARE) in their promoters [24-26]; many of these genes encode proteins involved in response to injury from oxidative stress [27].
The results in this study provide more insight into the transcriptional regulation of the HO-1 gene in human osteosarcoma cells and on the removal of intracellular ROS. These findings indicate that the nuclear translocation of transcription factor NFE2L2 play a crucial role in the transcriptional regulation of HO-1 gene, which was associated with the resistance of the cells to ATO. These findings also provide more understanding of the molecular mechanisms underlying the induction of HO-1 after exposure to ATO in osteosarcoma cells and are supported by the previous studies in which NFE2L2 was involved in inducing cisplatin resistance in ovarian carcinoma [28].
Conclusions
In the present study, we found that ATO up-regulated HO-1 expression in human osteosarcoma cells and that ATO activates HO-1 expression via its regulation of the transcription of the gene in MG63 cells. Furthermore, we determined that the activation of transcription was mediated by promoting the nuclear translocation of NFE2L2. These results indicate that transcriptional activation caused by the nuclear translocation of NFE2L2 is an underlying mechanism for the high induction of HO-1 which can account for the resistance of human osteosarcoma cells to ATO treatment.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grants: NSFC No. 21277057). We would like to express our great appreciation to Professor F. William Orr from the University of Manitoba in Canada for his great help in revising the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siclari VA, Qin L. Targeting the osteosarcoma cancer stem cell. J Orthop Surg Res. 2010;5:78-87
2. Ferrari S, Palmerini E. Adjuvant and neoadjuvant combination chemotherapy for osteogenic sarcoma. Curr Opin Oncol. 2007;19:341-6
3. Pakos EE, Nearchou AD, Grimer RJ, Koumoullis HD, Abudu A, Bramer JA. et al. Prognostic factors and outcomes for osteosarcoma: an international collaboration. Eur J Cancer. 2009;45:2367-75
4. Luetke A, Meyers PA, Lewis I, Juergens H. Osteosarcoma treatment - where do we stand? A state of the art review. Cancer Treat Rev. 2014;40:523-32 doi:10.1016/j.ctrv.2013.11.006
5. Emadi A, Gore SD. Arsenic trioxide - An old drug rediscovered. Blood Rev. 2010;24:191-9
6. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S. et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111-21
7. Mayor S. Arsenic trioxide combination improves survival in APL. Lancet Oncol. 2013;14:e346
8. Mi JQ, Li JM, Shen ZX, Chen SJ, Chen Z. How to manage acute promyelocytic leukemia. Leukemia. 2012;26:1743-51
9. Mann KK, Wallner B, Lossos IS, Miller WH Jr. Darinaparsin: a novel organic arsenical with promising anticancer activity. Expert Opin Investig Drugs. 2009;18:1727-34
10. Chow SK, Chan JY, Fung KP. Inhibition of cell proliferation and the action mechanisms of arsenic trioxide (As2O3) on human breast cancer cells. J Cell Biochem. 2004;93:173-87
11. Wang Y, Wei Y, Zhang H, Shi Y, Li Y, Li R. Arsenic trioxide induces apoptosis of p53 null osteosarcoma MG63 cells through the inhibition of catalase. Med Oncol. 2012;29:1328-34
12. Zhang H, Su X, Guo L, Zhong L, Li W, Yue Z. et al. Silencing SATB1 Inhibits the Malignant Phenotype and Increases Sensitivity of Human Osteosarcoma U2OS Cells to Arsenic Trioxide. Int J Med Sci. 2014;11:1262-9 doi:10.7150/ijms.10038
13. Andoh Y, Suzuki H, Araki M, Mizutani A, Ohashi T, Okumura T. et al. Low- and high-level expressions of heme oxygenase-1 in cultured cells under uninduced conditions. Biochem Biophys Res Commun. 2004;320:722-9 doi:10.1016/j.bbrc.2004.05.212
14. Maines MD, Gibbs PE. 30 some years of heme oxygenase: from a "molecular wrecking ball" to a "mesmerizing" trigger of cellular events. Biochem Biophys Res Commun. 2005;338:568-77 doi:10.1016/j.bbrc.2005.08.121
15. Barrera LN, Rushworth SA, Bowles KM, MacEwan DJ. Bortezomib induces heme oxygenase-1 expression in multiple myeloma. Cell Cycle. 2012;11:2248-52
16. Zhong L, Wang Y, Li W, Gu J, Li X, Wang X. et al. Heme oxygenase-1 silencing increases the sensitivity of human osteosarcoma MG63 cells to arsenic trioxide. Mol Cell Biochem. 2014;392:135-44 doi:10.1007/s11010-014-2027-1
17. Shi Y, Wei Y, Qu S, Wang Y, Li Y, Li R. Arsenic induces apoptosis of human umbilical vein endothelial cells through mitochondrial pathways. Cardiovasc Toxicol. 2010;10:153-60
18. Ramprasath T, Vasudevan V, Sasikumar S, Puhari SS, Saso L, Selvam GS. Regression of Oxidative Stress by Targeting eNOS and Nrf2/ARE Signaling: A Guided Drug Target for Cardiovascular Diseases. Curr Top Med Chem. 2015;15:857-71
19. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199-218 doi:10.1016/j.tibs.2014.02.002
20. Sanchez Y, Calle C, de Blas E, Aller P. Modulation of arsenic trioxide-induced apoptosis by genistein and functionally related agents in U937 human leukaemia cells. Regulation by ROS and mitogen-activated protein kinases. Chem Biol Interact. 2009;182:37-44
21. Wang Y, Xu Y, Wang H, Xue P, Li X, Li B. et al. Arsenic induces mitochondria-dependent apoptosis by reactive oxygen species generation rather than glutathione depletion in Chang human hepatocytes. Arch Toxicol. 2009;83:899-908
22. Das J, Ghosh J, Manna P, Sinha M, Sil PC. Taurine protects rat testes against NaAsO(2)-induced oxidative stress and apoptosis via mitochondrial dependent and independent pathways. Toxicol Lett. 2009;187:201-10
23. Gambari R. New trends in the development of transcription factor decoy (TFD) pharmacotherapy. Curr Drug Targets. 2004;5:419-30
24. Xu X, Zhang Y, Li W, Miao H, Zhang H, Zhou Y. et al. Wogonin reverses multi-drug resistance of human myelogenous leukemia K562/A02 cells via downregulation of MRP1 expression by inhibiting Nrf2/ARE signaling pathway. Biochem Pharmacol. 2014;92:220-34 doi:10.1016/j.bcp.2014.09.008
25. Cheng ZG, Zhang GD, Shi PQ, Du BS. Expression and antioxidation of Nrf2/ARE pathway in traumatic brain injury. Asian Pac J Trop Med. 2013;6:305-10 doi:10.1016/s1995-7645(13)60061-9
26. Mann GE, Niehueser-Saran J, Watson A, Gao L, Ishii T, de Winter P. et al. Nrf2/ARE regulated antioxidant gene expression in endothelial and smooth muscle cells in oxidative stress: implications for atherosclerosis and preeclampsia. Sheng Li Xue Bao. 2007;59:117-27
27. Yang JH, Shin BY, Han JY, Kim MG, Wi JE, Kim YW. et al. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol Appl Pharmacol. 2014;274:293-301 doi:10.1016/j.taap.2013.10.026
28. Bao LJ, Jaramillo MC, Zhang ZB, Zheng YX, Yao M, Zhang DD. et al. Nrf2 induces cisplatin resistance through activation of autophagy in ovarian carcinoma. Int J Clin Exp Pathol. 2014;7:1502-13
Author contact
![]() Corresponding authors: Dr. Ronggui Li, The Key Laboratory of Pathobiology, Ministry of Education, Norman Bethune College of Medicine, Jilin University, Changchun, 130021, P.R. China. Tel.: 86-431 85619481; Fax: 86-431-85619469; E-mail: lirgedu.cn and Dr. Zonggui Wang, The Second Hospital of Jilin University, Changchun, P.R. China. Tel.: 86-431 88796114; E-mail: zgw1965com.
Corresponding authors: Dr. Ronggui Li, The Key Laboratory of Pathobiology, Ministry of Education, Norman Bethune College of Medicine, Jilin University, Changchun, 130021, P.R. China. Tel.: 86-431 85619481; Fax: 86-431-85619469; E-mail: lirgedu.cn and Dr. Zonggui Wang, The Second Hospital of Jilin University, Changchun, P.R. China. Tel.: 86-431 88796114; E-mail: zgw1965com.
Received 2015-4-20
Accepted 2015-7-16
Published 2015-8-1