ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2015; 12(8):655-667. doi:10.7150/ijms.12460 This issue Cite
Research Paper
The Role of Autophagy in Kidney Inflammatory Injury via the NF-κB Route Induced by LPS
Yu Wu1,2, Yang Zhang3, Ling Wang2, Zongli Diao1, Wenhu Liu1 ![]()
1. Department of Nephrology, Beijing Friendship Hospital, Capital Medical University, No. 95 Yong An Road, Xi Cheng District, Beijing 100050, China
2. Department of Nephrology, The First People's Hospital of Xuzhou, No. 19 Zhongshan North Road, Xuzhou 221002, Jiangsu, China
3. Department of Anesthesiology, Xuzhou Medical College, Xuzhou 221004, Jiangsu, China
Abstract
Acute kidney injury (AKI) is a systemic inflammatory response syndrome associated with poor clinical outcomes. No treatments effective for AKI are currently available. Thus, there is an urgent need of development of treatments effective for AKI. Autophagy, an intracellular proteolytic system, is induced in renal cells during AKI. However, whether autophagy is protective or injurious for AKI needs to be clearly clarified. We addressed this question by pharmacological inhibition of autophagy using a mouse model of lipopolysaccharide (LPS) induced-AKI. We found that autophagy was induced in renal cortex of mice during LPS-induced AKI as reflected by a dose-and time-dependent increased accumulation of light chain 3-II (LC3-II), the common marker of autophagy, compared to that of control group; 2) the occurrence of intensive, punctate and increased immunohistochemical staining image of LC3-II in renal cortex; 3) the significant increase in the expression levels of Beclin-1, another key marker of autophagy; 4) the significantly increased levels of plasma urea and serum creatinine and 5) the significant increase in autophagagosome area ratio. We observed that 3-methyladenine (3-MA), a pharmacological inhibitor of autophagy, blocked autophagy flux, alleviated AKI and protected against LPS-induced AKI. LPS triggered kidney inflammation by activation of the canonical NF-κB pathway. This route can be modulated by autophagy. Activation of the canonical NF-κB pathway was reduced in 3-MA+LPS as compared to that in LPS-treated group of mice. Mice pretreated with 3-MA before exposure to LPS showed a reduction in p65 phosphorylation, resulting in the accumulation of ubiquitinated IκB. In conclusion, impairment of autophagy ameliorates LPS-induced inflammation and decreases kidney injury. The accumulation of ubiquitinated IκB may be responsible for this effect.
Keywords: autophagy, 3-methyladenine, inflammation, LPS-induced kidney injury, IκB
Introduction
Acute kidney injury (AKI), an abrupt loss of kidney function, is a systemic inflammatory response syndrome commonly occurring in critical patients. Its prevalence is 3-5% in patients with general hospital and can be as high as 30-50% in patient's intensive care unit [1]. Sepsis-induced AKI frequently occurs in the elderlies and is associated with poor clinical outcomes and high mortality [2-4]. However, as of to date, no effective treatment has been available for this devastating disease [5, 6]. While there are multiple clinical causes, the pathogenesis of AKI is primarily attributed to renal tubular sepsis damage [7]. Lipopolysaccharide (LPS), a bacterial endotoxin consisting of a lipid and a polysaccharide with O-antigen, elicits strong immune and inflammatory responses in animals. LPS challenge has been one of animal models commonly used to elucidate the mechanisms underlying sepsis-induced AKI and its potential treatment [8].
Autophagy is an intracellular degradation system by which the damaged proteins and dysfunctional organelles are delivered to autophagosomes and proteolytically processed there [9]. During autophagy, microtubule-associated protein 1A/1B-light chain 3 (LC3) was initially modified by the lipidation. These lipidated LC3 molecules, known as LC3-II, are the key components constitutively present in the membrane of autophagosome. Inhibition of autophagy leads to a reduced level of LC3-II isoforms. Autophagy has also been increasingly implicated to play an essential role in the regulating both pro- and anti-inflammatory responses [10]. While autophagy has been regarded as a survival mechanism, abnormal (e.g. excessive) autophagy may result in cell death [11].
Autophagy has been implicated in the pathogenesis of a variety of diseases including heart failure, cancer, neurodegenerative diseases, and other diseases [12]. In kidneys, autophagy has been suggested to play an essential role in maintaining homeostasis and physiological functions [13]. However, its precise roles in the pathogenesis of AKI still need to be clearly defined, although several studies have suggested that autophagy may play a renoprotective role in AKI [14-17] and a role in regulation of tubular cell death [18-21]. This study aimed to define the roles of the autophagy in responding to the adverse effects induced by LPS. We compared the severity of kidney injury in normal mice, mice exposed to LPS and mice pretreated with 3-methyladenine (3-MA), an autophagy inhibitor [22], followed by LPS-challenge. By investigating the differences in the severity of kidney injury among these groups of mice, we should be able to clarify whether autophagy is an adaptive/protective or a pathogenic mechanism for AKI.
Materials and Methods
Animals
Wild-type C57BL/6J male mice (10-14 weeks old) were purchased from the Experimental Animal Centre of Xuzhou Medical College (Xuzhou, Jiangsu, China). Mice were handled under a protocol approved by the Animal Care and Use Committee of the Xuzhou Medical College (Approval ID: SCXK-Su 2010-0003). These mice were maintained under specific pathogen-free (SPF) conditions, and provided with a 12-h/12-h light/dark cycle and free access to both food and water. The temperature and relative humidity within the animal room were maintained at 22-25°C and 40-60%, respectively, for 1 week before being used for the experiments.
Experimental protocols
A total of 32 male mice were randomly divided into four groups with 8 mice per group. The mice were administrated with LPS (Cell Signaling, Beverly, MA, USA) at 10 mg/kg body weight (BW) to induced endotoxemia as described [23]. Briefly, the mice in the first group were given an intraperitoneal (I.P.) injection with 0.9 % normal physiological saline (NPS) and used as the controls (designated as CON)(n=8); The mice in the second group were administrated with a single I.P. injection of 3-MA (Cell Signaling) at 15 mg/kg (in 0.1 mL of 0.9 % NPS) (n=8)(designated as 3-MA). The mice in the third group were administrated with a single I.P. injection of LPS (10 mg/kg in 0.1 mL of 0.9% NPS)(n=8)(designated as LPS); and the mice in the fourth group were pretreated intraperitoneally with 3-MA at 15 mg/kg (in 0.1 mL of 0.9 % NPS)(n=8) for 1 h, followed by a challenge with LPS (10 mg/kg in 0.1 mL of 0.9 % NPS n=8)(designated as LPS+3MA) as described [24]. At 24 after IP-injection, the physical activities of some mice were decreased slightly but no mice showed the weight loss, poor body, and abnormal skin during the treatment. After 24 hours, a ketamine/xylazine mixture (75 mg/kg) was intraperitoneally injected into each mouse of all the groups to anesthetize them. All the animal experiments were carried out at body temperature (37 and 38℃), which was maintained with a heating lamp. Blood samples were collected and the serum creatinine levels and plasma urea levels were examined. Thereafter, all the mice were euthanized and the injury to their kidney tissue was assessed by both histological and biochemical analyses.
Biochemical Examination
The levels of the serum creatinine and plasma urea were measured using an Olympus AU2700 automatic biochemistry apparatus (Olympus America Inc., Melville, NY, USA).
Western Blotting Analysis
The total protein was extracted from kidney tissues as described [25]. Total protein (100 μg/well) was firstly separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and then immunobloted to the nitrocellulose membranes according to the instructions given by the manufacturer (Bio-Rad, Hercules, CA, USA). The nitrocellulose membranes were blocked with 5% non-fat dry milk in TBST buffer (10 mmol/l Tris-HCl, 0.15 mol/l NaCl and 0.05% Tween 20, pH 7.2) for 1 h and then incubated with antibodies against LC3 (Sigma), Beclin-1 (Abcam, Cambridge, UK); phosphorylated p65 (Abcam), p52(Cell Signaling), and IL-1β (Santa Cruz Biotechnology), respectively, at 4oC overnight, washed and then incubated with the corresponding goat anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (Santa Cruz Biotechnology) in 1:3,000-5,000 (in PBST) for 60 min. Protein bands were developed and detected using the ECL Super Signal reagent (Pierce, Rockford, IL, USA). Relative band densities of the indicated target proteins were measured from scanned films using NIH ImageJ Software.
Immunohistochemical Staining
After being fixed with 10% neutral buffered formalin for 24 h, the renal tissues were embedded in paraffin and sectioned at 4 um according to the standard procedure. The sections were deparaffinized, hydrated gradually, and stained immunohistochemically as described previously [25]. The procedures included microwave antigen retrieval (in citrate buffer, 0.01 mol/l, pH 6.0). Endogenous peroxidase was blocked with 3% H2O2 for 15 min. The sections were firstly blocked with 4% goat serum to minimize the non-specific staining and then stained with affinity-purified polyclonal rabbit anti-LC3 antibody (Sigma, St. Louise, MO, USA) diluted into 1:100 in PBST (PBS, pH 7.4, 0.05% Tween 20). The sections were incubated at 4oC overnight. The bound primary antibody was detected by horseradish peroxidase conjugated anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and developed with 3,3-diaminobenzidine tetrahydrochloride. Finally, the expression levels of the immunochemically stained LC3 protein were analyzed and evaluated by the average optical density (AOD) and integral optical density (IOD) of staining in 200X magnification under microscopic examination[25] (Olympus, Tokyo, Japan).
Visualization of Renal Tissues with Transmission Electron Microscopy (TEM)
After being excised, the kidney tissues were fixed with a fixative buffer (2% paraformaldehyde and 2.5% glutaraldehyde in 0.1M of phosphate-buffered solution) and stored at 4°C before being embedded. Tissue samples were then postfixed in 1% phosphate-buffered osmium tetroxide and embedded in Spurr's resin. Ultrathin sections (0.1 μm) were made, stained consecutively with 1% uranyl acetate and 0.2% lead citrate, and visualized with TEM (JEM-1220). Using Adobe Photoshop CS3 Extended software, the total autophagosomal areas,and the percentage of the autophagosome-occupied cells were measured, calculated and expressed as autophagosome area ratio (%) as described previously [24].
Immunoprecipitation
Immunoprecipitation analysis was performed as described previously [24] as follows: approximately 300 μg of kidney tissue protein at 4°C was immuno-precipitated with 1 μl of rabbit anti-IκB antibody (Cell Signaling) at 4°C for 90 min, followed by adding Protein G Plus-Agarose (Santa Cruz Biotechnology)., The mixture was incubated overnight and centrifuged. The supernatants were discarded. The pelleted immunocomplexes were denatured by heating at 99°C for 5 min, loaded into the well, separated on 13% SDS-PAGE and analyzed by Western blotting with both anti-polyubiquitin (FK1, EnzoLife Sciences, Farmingdale, NY, USA), and anti-p65 (Abcam) antibodies, respectively.
Statistical Analysis
The differences between control and the experimental groups were determined by using One-way ANOVA and Student's Newman-Keuls test for post-hoc comparisons. Student's t-test was conducted for paired samples. The differences in the changes of the parameter examined over time between different groups were evaluated by a two-way ANOVA with repeated measures. Data were expressed as mean ± SEM, and the differences between group means with P < 0.05 were considered statistically significant.
Results
Activation of autophagy induced by LPS stimulation
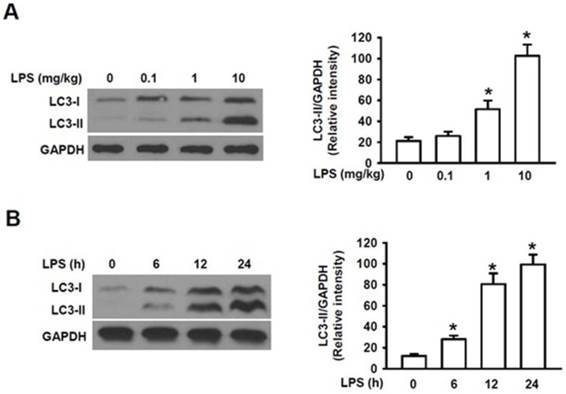
To elucidate the mechanism by which autophagy plays the roles in the mechanism of AKI, we first determined whether LPS can cause activation of autophagy in the kidney. Microtubule-associated protein 1 light chain 3 (LC3) is a 16 kDa soluble protein present ubiquitously in mammalian cells and plays a critical role in the macroautophagic formation. It has been used a common marker for autophagy [26]. When autophagy is induced, the newly synthesized LC3 precursor is firstly cleaved by Atg4B, a human cysteine protease, to generate LC3-I in cytosol, which is then converted to the membrane-bound LC3-II by adding phosphatidylethanolamine (PE) to glycine residue 120 at its C-terminal. LC3-II is firmly bound to the membrane of autophagosome and is, thus, regarded as a specific marker for autophagy [26]. Fig. 1A showed that challenge of male mice with LPS at the doses of 0.1, 1.0 and 10 mg/kg for 24 h induced a dose-dependent, gradual increase in the accumulation of LC3-II as compared to that of the control group (0 mg/kg) and a markedly increased accumulation level of LC3-II was induced at 10 mg/kg compared with those at 0.1 and 1 mg/kg. The male mice were then challenged with LPS at 10 mg/kg for 0, 6, 12 and 24 h, respectively. We also observed that the levels of LC3-II in the kidney were significantly increased within 6h following LPS stimulation and reached to the remarkably high levels within 24 h (Fig. 1B). Fig. 1A and 1B also showed that the increased accumulation of LC3-I were followed by the subsequently increased accumulation of the LC3-II in both dose- and time-dependent manners, indicating the increased conversion of LC3-1 into LC3-II induced by LPS stimulation. These results clearly indicate that challenge of mice with LPS markedly induces LC3I expression and its subsequent cleavage into LC3II in a dose- and time-dependent manner and induces activation of autophagy in the mouse kidney.
Induction of Autophagy mainly occurred in renal cortex during LPS-induced AKI in mice
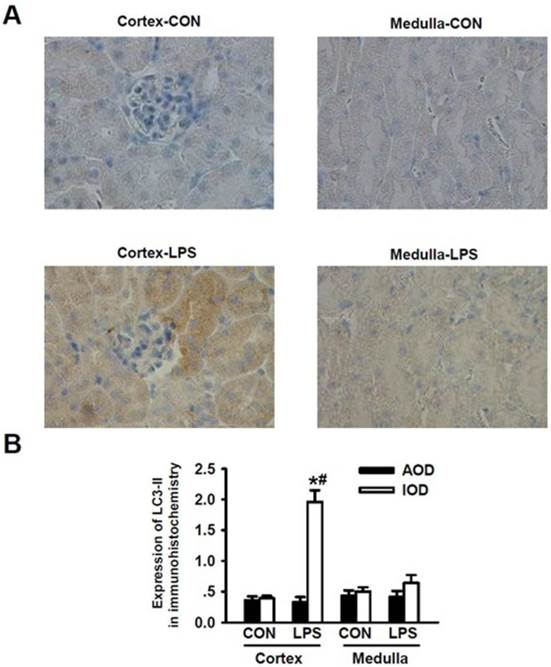
According to the unique structural and functional features of kidney, renal cortex plays roles in filtrating blood and forming crude urine, thus, a large number of renal cortexes are present in the glomerulus while kidney medulla plays an important role in re-absorption and concentration of urine and thus, a large number of distal renal tubules are present in kidney medulla but there are no glomerulus there. We applied immunohistochemical staining method to detect the LPS-induced expression of LC3-II in kidney tissue. We visualized and analyzed the expression levels of LC3-II in renal cortex and medulla, respectively. As shown in Fig. 2A, after being stimulated with LPS, the immunohistochemical staining intensity for LC3-II in renal cortex was significantly enhanced as compared to that of LC3-II in renal cortex of the normal mice (Fig. 2A, panel cortex-LPS versus panels Cortex-Con); Morphologically, the formation of autophagosomes in kidneys was visualized by immunohistochemical staining of LC3-II. In kidney tissues of the controlled mice, LC3-II was diffusely distributed throughout the cells without punctate staining. Upon LPS stimulation for 24 h, intensive, punctate and increased LC3-II staining appeared mainly in renal cortex, indicating the formation of autophagosomes there. The immunohistochemical staining intensity of LC3-II in renal medulla was not obviously increased (Fig. 2A, panel Medulla-LPS versus panel Medulla-Con). Quantitative analysis of the immunohistochemical staining image intensity also showed the significant increase in LC3-II staining in renal cortex of mice after being stimulated with LPS. We found that the immunohistochemical staining intensity of LC3-II was higher in renal cortex than in renal medulla (Fig. 2B). These lines of compelling evidence clearly demonstrate that the occurrence of autophagy is induced in kidney renal cortex during LPS-induced AKI.
Analysis of the expression of microtubule-associated protein LC3-II in kidney homogenate by Western blot. A. The expression levels of LC3-II proteins in kidney homogenates of male mice intraperitoneally injected (I.P.) with lipopolysaccharide (LPS) at the indicated doses (n=6 for each dose). The assay was repeated three times. Left panel, the representative Western blots for LC3-II; Right panel, quantification of LC3-II by densitometry (n=3); *P<0.05 vs 0 mg/kgLPS group; B. The expression levels of LC3-II proteins in kidney homogenates of male mice intraperitoneally injected with lipopolysaccharide (LPS) at 10 mg/kg for the indicated time points (n=6, for each time points). Left panel, the representative Western blots for LC3II; Right panel, protein quantification of LC3-II by densitometry (n=3); *P<0.05 vs. LPS stimulation at 0 h.
Detection of LC3-II expressions for the occurrence of LPS-induced autophagy in renal cortex and medulla via immunohistochemical staining. A. Immuohistochemical staining of LC3-II in renal cortex and medulla of mice intraperitoneally injected without (upper panels) and with (lower panels) lipopolysaccharide (LPS) for 24 h: compared to those in the cortex (cortex-con) and medulla (medulla-con) of the controlled mice, the immunohistochemical staining intensity of LC3-II was significantly increased in cortex (cortex-LPS) but only slight increase in medulla (medulla-LPS) of the LPS-stimulated mice. B. Analysis of the average optical intensity (AOD) and integral optical density (IOD) of the immunohistochemical staining intensity of LC3-II in cortex and medulla of mice stimulated without or with LPS. The immunohistochemical staining intensity of LC3-II in renal cortex was significantly increased in LPS-stimulated mice (Cortex-LPS), *P<0.05 vs. Cortex-CON (n=8); while the immunohistochemical staining intensity of LC3-II in renal cortex of LPS-stimulated mice was also significantly higher than in renal medulla (Medulla-LPS), # P<0.05 vs. Medulla-LPS (n=8).
Inhibition of autophagy by 3-MA dramatically attenuated LPS-induced AKI in mice
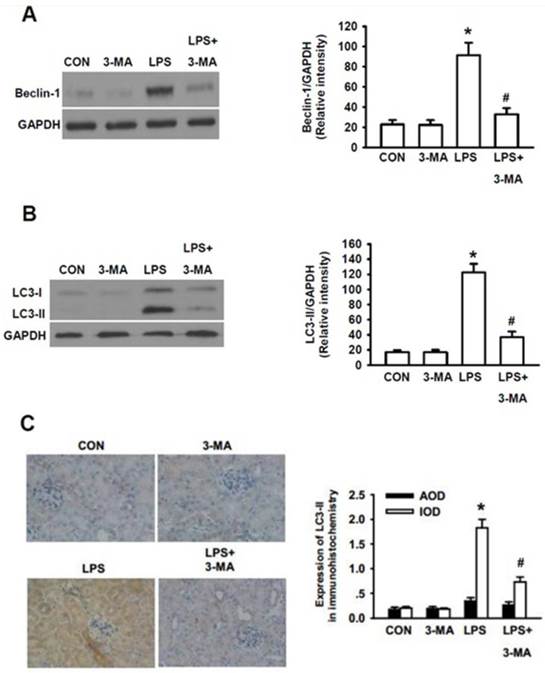
Since the precise roles of autophagy in the pathogenesis of AKI still remain controversial, we next determined whether or not autophagy actually plays an essential role in the pathogenesis of AKI. We initially tested the effects of 3-methyladenine (3-MA), a pharmacological inhibitor of autophagy, on LPS-induced AKI in mice. It has been demonstrated that 3-MA is capable of blocking the formation of autophagosome, leading to inhibition of autophagic activation [27]. Thus, we addressed whether 3-MA inhibited the LPS-induced autophagic activation by examining whether 3-MA could inhibit LPS-induced expression of Beclin-1, another commonly used biomarker for autophagy [28]. As shown in Fig. 3A, 3-MA itself did not cause effects on the expression level of Beclin-1 as compared to that of the control group. Significantly higher level of Beclin-1 was seen in LPS-stimulated group (P<0.05) and 3-MA almost completely blocked the LPS-induced accumulation of Beclin-1 (P<0.05). Similarly, significantly higher levels of LC3-II were induced by LPS-stimulation (P<0.05) and 3-MA almost completely blocked the LPS-induced accumulation of LC3-II (P<0.05)(Fig. 3B). Consistent with the Western blot results, quantitative analysis of immunohitochemical staining of LC3-II revealed that integrated optical density (IOD) of LC3II staining was significantly weaker in the 3-MA-pretreated mice than in the mice induced only by LPS whereas bare LC3 dot was observed either in the control or in the LPS+3-MA group mice (Fig. 3C).
Inhibition of autophagy by 3-MA dramatically reduced the severity of LPS-induced AKI
Inhibition of autophagy by 3-MA dramatically attenuated LPS-induced AKI in mice. The mice were pretreated i.p. without or with 3-MA (15 mg/kg in 0.1 mL of 0.9 % normal saline)(n=8) for 1h, followed by exposure to LPS (10 mg/kg in 0.1 mL of 0.9 % normal saline)(n=8) for 24h. The homogenates of one side of kidney were used to examined the expression levels of Beclin-1(A) and LC3-II (B). The other side of kidney was used for immunohistochemical staining for LC3-II to examine its expression in renal cortex (C). A. Left panel, representative Western blots for Beclin-1; Right panel, quantification of Beclin-1 protein by densitometry (n=3); B. Left panel, the representative Western blots for LC3-II; Right panel, quantification of LC3-II protein by densitometry (n=3); C. left panel, immunohistochemical staining for LC3-II protein in renal cortex; right panel, quantitative analysis of the optical intensity of the immunohistochemical staining image of LC3-II protein. *P<0.05 vs. CON; # P<0.05 vs. LPS.
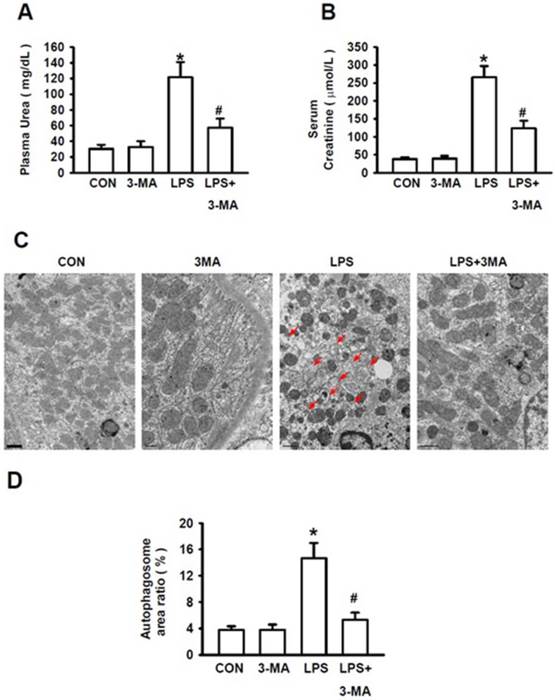
We then examined the severity of LPS-induced AKI in the absence or presence of 3-MA. The ratio of blood urea nitrogen to blood creatinine was regarded as a prognostic indicator of mortality [29]. Stimulation of mice with LPS at 10 mg/kg caused the significantly increased levels of plasma urea (Fig. 4A) and serum creatinine (Fig. 4B), indicating a more severe AKI induced by LPS. However, the levels of plasma urea and serum creatinine were not affected by 3-MA alone as compared to those of the control group. Mice pretreated with 3-MA for 1h, followed by LPS stimulation, displayed significantly lower levels of plasma urea (Fig. 4A) and serum creatinine (Fig. 4B). Consistent with these biochemical indexes for autophagy, visualization of renal tissues with TEM clearly revealed that treatment of mice with 3-MA alone did not increase the autophagosome area ratio (%) as compared to that of the control group whereas stimulation of mice with LPS caused a significant increase in the autophagosome area ratio but pretreatment of mice with 3-MA almost completely inhibited LPS-induced increase in autophagosome area ratio (Fig. 4C and 4D)(P<0.05). Visualization with TEM revealed a number of pathological changes that had occurred within the renal cortex cells of mice challenged with LPS, i.e. their mitochondria became shrunk and fractured, and their mitochondrial cristae were defective and loss. The number of organelles was reduced and a large number of empty vacuoles and residue bodies appeared. These pathological changes were significantly ameliorated when the LPS-induced autophagy was inhibited by 3-MA (Fig. 4C). Collectively, these results strongly demonstrate that selective inhibition of autophagic activation by pharmacological inhibitor has a protective effect against AKI in this experimental model, suggesting that the pharmacological agents, such as 3-MA, that can modulate autophagy, might be valuable for treatment of AKI.
Pharmacological inhibition of autophagy improved kidney injury. The mice were pretreated i.p. without or with or 3-MA at 15 mg/kg (in 0.1 mL of 0.9 % normal saline)(n=8) for 1h, followed by exposure to LPS (10 mg/kg in 0.1 mL of 0.9 % normal saline, n=8) for 24h. Blood samples were collected from mice in these groups. The levels of the serum creatinine and plasma urea were measured. A. Levels of plasma urea; B. Levels of serum creatinine; C. TEM visualization of the injury in renal cortex of mice, the red arrows point to autophygosomes; D. Quantitative analysis of the number per area of autophygosomes; *P<0.05 vs. CON; #P<0.05 vs. LPS.
Impairment of autophagy resulted in defective NF-κB activation and accumulation of ubiquitinated IκB
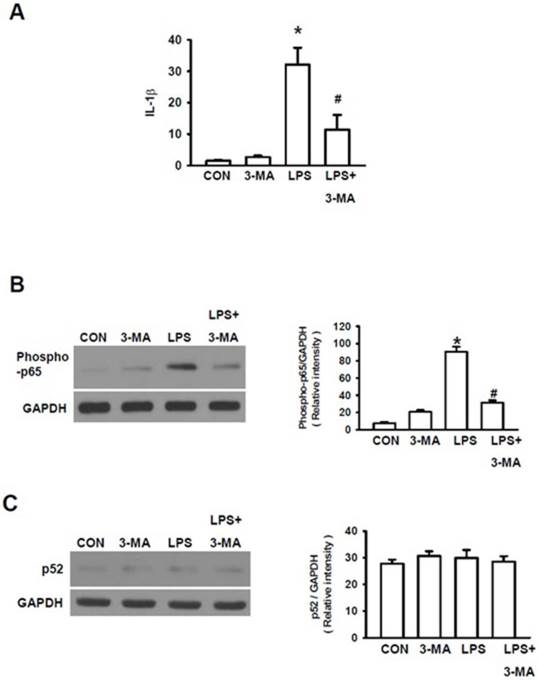
As it has been documented that various inflammatory cascades are involved in the development of kidney dysfunctions [30] and that LPS can trigger AKI via inflammatory injury [31], we investigated the roles of inflammatory cascades in LPS-induced AKI. Firstly, we measured the expression of interleukin-1β (IL-1β) gene, one of the primary pro-inflammatory mediators. LPS significantly increased the expression level of IL-1 gene (Fig. 5A). Since IL-1β can activate the nuclear factor NF-κB pathway to amplify the inflammatory response, we further examined the effects of LPS on stimulation of NF-κB pathway, a key prototypical proinflammatory signaling pathway [32]. NF-κB pathway has been subdivided into classic (canonical) and alternative (non-canonical) NF-κB pathway [32]. The canonical NF-κB pathway is mainly responsible for responding to TNFα and IL-1 signaling, which plays an important role in the pathogenesis of chronic inflammatory diseases. NF-κB is primarily activated via IκB kinase-mediated phosphorylation by inhibitory molecules, including IκBa. Phosphorylation of proteins involved in NF-κB pathway, including p65, is also required for optimal induction of NF-κB target genes [33]. On the other hand, the alternative (non-canonical) NF-κB pathway is involved in the inducible phosphorylation of p100 by IKKα, which, in turn, activates RelB/p52 heterodimers [32]. Thus, we examined the effects of LPS-stimulation on both p65 phosphorylation and p52 phosphorylation to determine whether canonical pathway or non-canonical pathway or both are involved in LPS-induced AKI. LPS caused p65 phosphorylation (Fig. 5B), resulting in the activation of the canonical NF-κB pathway. However, mice pretreated with 3-MA followed by LPS stimulation displayed a significant decrease in IL-1 gene expression (Fig. 5A) and p65 phosphorylation (Fig. 5B) as compared to those in mice stimulated with LPS alone. However, no significant differences in p52 phosphorylation were seen between these groups of mice, as measured by Western blot using the p52 antibody (Fig. 5C), suggesting that the canonical NF-κB pathway but not the non-canonical route of NF-κB pathway is involved in 3-AM inhibition of LPS-induced autophagy.
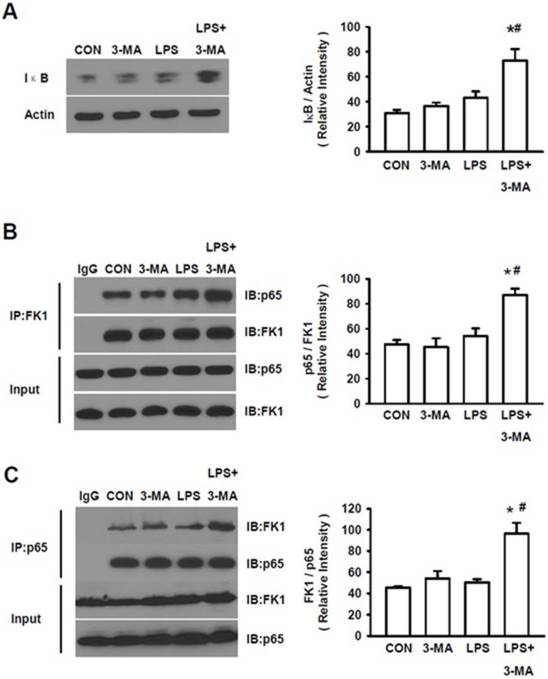
Ubiquitination/processing of IκB has been known to be an essential step for NF-κB activation [32, 33]. As shown above, mice pretreated with 3-MA displayed the increased levels of an ubiquitinated protein, LC3-II, we then analyzed the accumulation of IκB. A significant increase in total IκB levels was seen in kidney of mice pretreated with 3MA followed by LPS challenge (Fig. 6A). We also measured ubiquitination of IκB after co-immunoprecipitation with either antibody for p65 or with the anti-polyubiquitylated conjugates mAb FK1. There was an increase in ubiquitinated IκB in 3-MA pretreated animals challenged by LPS (Fig. 6B). Moreover, p65 levels were increased in co-immunoprecipitation, as evident by the co-immunoprecipitation with antibody for p65 (Fig. 6B) and with FK1 (Fig. 6C), demonstrating the binding of this protein to IκB. These results show that inhibition of autophagy leads to the accumulation of ubiquitinated IκB that is bound to p65, preventing the activation of the canonical NF-κB pathway.
Autophagy activated NF-κB pathway. A. Measurement of the expression level of IL-1β gene To characterize which the intracellular proinflammatory pathways are involved in LPS-induced autophagy, both canonical and non-canonical routes of NF-κB were studied with Western blot. B. LPS stimulated the classic canonical route via phosphorylation of p65; C. but did not cause any effects non-canonical route as indicated by without changes in p52 phosphorylation : * P<0.05 vs. CON; #P<0.05 vs. LPS.
Accumulation of ubiquitylated IκB. A. The total IκB levels were measured in kidney tissue homogenates of mice in four groups. Mice pretreated with 3MA (15 mg/kg) followed by challenge with by LPS (10 mg/kg) displayed significantly increased levels of IκB. After being co-immunoprecipitated with either antibody for p65 or with FK1, the LPS+3MA-treated mice displayed significantly higher levels of p65 (B) and FK1(C), demonstrating the binding of both ubiquitin and p65 to IκB in treated animals. *P<0.05 vs. CON; #P<0.05 vs. LPS.
Discussion
As a major kidney disease frequently occurring in elderly patients, AKI is associated with poor clinical outcomes and high mortality. Currently, no treatment effective for AKI is available. Several studies have reported that autophagy is induced in renal tubular cells during AKI where it may play certain roles in protection against kidney injury [14, 34-36]. However, the precise mechanisms underlying its protective effects against AKI need to be determined. Elucidation of the precise mechanisms underlying its protective effects against AKI will be valuable for development of treatment effective for AKI. In this study, we addressed this key issue and made some important observations.
Our finding that autophagy is induced in renal cortex during LPS-induced AKI in mice. This finding is consistent with the previous observations that autophagy was induced in proximal tubules during renal ischemia-reperfusion (I/R) in mice [14] and cisplatin-induced AKI in C57BL/6 mice [34, 36].
While multiple factors are involved in its pathogenesis, the pathogenesis of ARI is mainly related to renal tubular injury and cell death there [34]. The renal cortex, which includes both renal tubules and renal corpuscles, is the outer portion of the kidney where ultrafiltration takes place. The proximal tubule is the portion of the duct system of the nephron in the kidney. The severity of kidney injury and the associated pathophysiological changes are related to the nature and the frequency of the insults [37]. For instance, it was reported that the cisplatin-induced AKI was associated with the pathophysiological changes including proximal tubular injury, inflammation, activation of multiple proinflammatory cytokines, infiltration of inflammatory cells and apoptosis in the kidney [38]. Jian et al. observed that in mice whose Atg7 gene, which encodes an E1-like activating enzyme involved in autophagy, was specifically knocked out, the proximal tubular cells isolated from their kidney tissues were more sensitive to renal I/R-induced injury and cisplatin-induced apoptosis as compared to those in kidney tissues isolated from the wild-type mice [14]. Similarly, Kimura et al. reported that elimination of autophagy in the proximal tubule by specifically knocking out Atg5 gene, which encodes an E3 ubiquitin ligase essential for autophagy, caused substantial accumulation of deformed mitochondria, cytoplasmic inclusions, cellular hypertrophy and degeneration [39]. In agreement with this observation, we observed that within the renal cortex cells of mice challenged with LPS, their mitochondria became shrunk and fractured, and the mitochondrial cristae were defective and loss, indicating that in addition to autophagy, mitophagy is also induced in proximal tubule by LPS-induced AKI. Together, these observations clearly demonstrate that both autophagy and mitophagy are activated in proximal tubule and may play a role in protecting proximal tubule from degeneration and I/R-induced- and LPS-induced kidney injury.
In this study, we found that challenge of mice with LPS substantially induced the expression of IL-1β, and that this LPS-induced IL-1β expression was significantly inhibited by 3-MA. Consistent with this observation, it has been reported that tumor necrosis factor-α (TNF-α), another pro-inflammatory cytokine which acts directly on TNF receptor-1 in kidney, plays a key role in LPS-induced acute renal failure (ARF)[40]. These results support the notion that autophagy is involved in modulation of LPS-induced inflammatory responses. In fact, AKI is a systemic inflammatory response syndrome. Increasing evidence indicates that autophagy can plays multiple roles in modulating the inflammatory responses. Autophagy is involved in the negative regulation of p62, a signaling adaptor implicated in the activation of NF-κB [41]. Autophagy has also been demonstrated to play an anti-inflammatory role [42, 43]. For instance, Nakahira et al. [42] reported that in mice whose LC3-II and Beclin-1 were knocked out, dysfunctional mitochondria and cytosolic translocation of mitochondrial DNA were substantially accumulated, which led to the activation of caspase-1 and secretion of IL-1β and IL-18. LC3-II-knocked out mice exhibited higher susceptibility to LPS-induced mortality. These observations suggest that autophagic proteins are involved in inhibition of inflammation by preserving mitochondrial integrity. This is in agreement with our observation that the mitochondria in renal cortex cells of LPS-challenged mice became shrunk and fractured, and their mitochondrial cristae were defective and loss and that these LPS-induced alterations in mitochondrial morphology and structure was ameliorated by inhibition of autophagy with 3-MA. Saitoh et al [43] demonstrated that autophagy-related 16-like 1 (Atg16L1), an essential component of the autophagic system, was involved in regulation of LPS-induced activation of inflammasome in mice. The levels of both IL-1β and IL-18 in macrophages of Atg16L1-deficient mice were increased. Challenge of Atg16L1-knocked out mice with LPS caused caspase-1 activation and the increased IL-1β expression in their macrophages. Thus, these studies suggest that autophagy may be a beneficial mechanism that removes damaged organelles. However, on the other hand, other studies have shown that when the autophagy was blocked, the decreased inflammatory responses might be mediated through different mechanisms. Among which, the most notable one is the proteolytic processing of IκB, which is required for the activation of NF-κB and can occur in both the proteasome and the autophagosomes [44, 45]. The proteolytic pathway is blocked when autophagy is targeted. It was reported that targeting the essential molecules of the autophagic pathway inhibited the cellular response to TNF-α [46]. Thus, we hypothesized that the amplification of LPS-triggered inflammatory responses can be limited through inhibiting autophagy. The accumulation of ubiquitinated IκB observed in our study may be explained by the crosstalk between autophagy and the ubiquitin-proteasome pathway although whether these results are caused by inhibition of autophagy itself or by the impaired proteosomal delivery or by both can't distinguished at the present study.
As an inhibitor, 3-MA has been used to inhibit the formation of autophagosome [23]. Autophagy plays an essential role in maintaining the quality of cellular constituents by continuously recycling the damaged protein and/or the entire organelles to lysosome for degradation and processing [47]. Jiang et al. reported that blockage of autophagic flux with chloroquine, another inhibitor of autophagy, enhanced AKI whereas autophagic activation with Rapamycin, an inhibitor of mTOR that promotes autophagy, protected against cisplatin-induced and I/R-induced AKI in mouse models [14]. Induction of autophagy may be helpful to preserve homeostasis in response to stresses. Enhanced mitophagy, which is responsible for clearance of the damaged mitochondria, may also contribute to the improvement of cell viability [48]. In addition to autophagy and mitophagy, pexophagy may also be also involved in AKI. However, until to date, no study has been done on the role of pexophagy in LPS-induced renal injury and very few studies have defined the roles of peroxisomes and pexophagy in organ damage. Thus, the precise and complete mechanisms underlying the connections between inflammatory and autophagy networks are still far from being understood [49, 50]. Furthermore, it is also possible that the NF-κB activation itself is a trigger of autophagy as described in cell survival after heat shock in the in vitro experimental models [51].
Our results indicated that AKI induced by LPS-challenge was potently inhibited by 3-MA, suggesting that pharmacological modulation of autophagy during this kidney injury could be beneficial for treating AKI. This notion is also supported by several previous studies. For instance, Kakahashi et al. [52] reported that LPS-induced systemic inflammatory response syndrome (SIRS) was ameliorated by fidarestat, an aldose reductase inhibitor, resulting in the lowered mortality. Similarly, the autophagy-diminished older adult mice with LPS-induced AKI were unable to recover their renal function. Administration of temsirolimus, the mTOR inhibitor, induced autophagy and protected against AKI even after endotoxemia was established [53]. Xu et al. [54] reported that Leonuring (LEO), a major bioactive component from Herbal Leonuri, protected against LPS-induced AKI, improved animal survival by maintaining the redox balance and that these beneficial effects were attributed to its down-regulation of the expression of a number of proinflammatory cytokines and inhibition of both IκBα phosphorylation and p65 translocalization. Zhang et al. [55] reported that LPS-induced AKI was ameliorated by paclitaxel, a microtubule-stabilizing agent with anticancer activity, through its binding to myeloid differentiation protein-2 (MD-2), leading to blockage of the activation of NF-κB and cytokine production. Sun et al. [11] reported that in a model of acute lung injury caused by H5N1 virus, inhibition of autophagy improved outcome by reducing cell death. Non-injurious ventilation also caused autophagy-mediated effects, including diaphragm atrophy [56] which could be beneficial from inhibiting this pathway. However, like with any other treatments, caution must be taken with this approach. Since autophagy is an adaptive mechanism responsible for enhancing cell survival, any inappropriate interference may cause detrimental effects. An increased susceptibility to hypoxia-induced pulmonary hypertension has been associated with the lack of LC3 [57]. Furthermore, absence of autophagy could be favorable for the cytoplasmic accumulation of aggregates of the damaged proteins and/or deformed mitochondria [58], resulting in tissue dysfunction. Finally, the proinflammatory effects of targeting autophagy in sepsis [44] could also cause the adverse effects as well. While the clinical relevance and significance of modulation of autophagy in patients with kidney injury remain to be fully elucidated, the contribution of autophagy to inhibition of AKI and the potential benefits of its inhibition can be a promising therapeutic method that deserves attention and development.
It has been demonstrated that the impairment of autophagy result in less severity of injury, which is attributed to the inhibition of the activation of NF-κB pathway [45]. It is likely that the accumulation of ubiquitinated proteins, including IκB, could be associated with the inhibition of autophagy to the decreased NF-κB activation [45]. We found that autophagy in kidney is mediated mainly via the classic NF-κB pathway but not via the non-classic alternative NF-κB pathway. Ubiquitination and subsequent processing of the ubiquitinated IκB is the essential step during the activation of NF-κB pathway as the increased accumulation levels of ubiquitinated proteins were seen as IκB-knockout mice [59]. Chen et al. (60) monitored the changes of IkkB during acute I/R-induced injury and ischemic preconditioning (IPC), a short period of sub-lethal ischemia preceding ischemic myocardium which protects ischemic myocardium. They found that renal I/R-induced injury could be attenuated by IPC through inhibiting IkkB activation and inflammatory responses.
Our results have clearly shown that impairment and/or inhibition of autophagy can lead to the increased accumulation of ubiquitinated IκB that is bound to p65 and blockage of the NF-κB pathway, which can ameliorate LPS-induced AKI. These results indicate that impairment/inhibition of autophagy with 3-MA ameliorated LPS-induced AKI by preventing the activation of the classic canonical NF-κB pathway
Conclusions
In summary, our results have shown that autophagy is induced in renal cortex during LPS-induced AKI and that inhibition of autophagy decreases kidney injury after LPS stimulation by preventing the activation of canonical NF-κB pathway. The inhibition of IκB degradation could be one of the mechanisms responsible for this effect. This study opens the possibility to prevent LPS-induced AKI by the inhibition of the autophagic responses.
Acknowledgements
This study received financial support from the National Natural Science Foundation of China (81300607), Beijing Municipal Science and Technology Commission Funds (D131100004713001), and the National Key Technology R&D Program (2011BAI10B08).
Competing Interests
The authors have declared that no competing interest exists.
References
1. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. Kidney Int. 2012(Suppl 2):19-36
2. Suh SH, Kim CS, Choi JS. et al. Acute kidney injury in patients with sepsis and septic shock: risk factors and clinical outcomes. Yonsei Med J. 2013;54:965-72
3. Maddens B, Vandendriessche B, Demon D. et al. Severity of sepsis-induced acute kidney injury in a novel mouse model is age dependent. Crit Care Med. 2012;40:2638-46
4. Lim JH, Kim EN, Kim MY. et al. Age-associated molecular changes in the kidney in aged mice. Oxid Med Cell Longev. 2012;2012:171383
5. Nguyen HB, Rivers EP, Abrahamian FM. et al. Severe sepsis and septic shock: review of the literature and emergency department management guidelines. Ann Emerg Med. 2006;48:28-54
6. Venkataraman R, Kellum JA. Sepsis: update in the management. Adv Chronic Kidney Dis. 2013;20:6-13
7. Wang Z, Holthoff JH, Seely KA. et al. Development of oxidative stress in the peritubular capillary microenvironment mediates sepsis-induced renal microcirculatory failure and acute kidney injury. Am J Pathol. 2012;180:505-16
8. McCurdy TR, Patrick AL, Eltringham-Smith LJ. et al. Alpha-1 acid glycoprotein reduces hepatic leukocyte recruitment in murine models of either early endotoxemia or early sepsis. Microcirculation. 2014;21:74-83
9. Marino G, Lopez-Otin C. Autophagy: molecular mechanisms, physiological functions and relevance in human pathology. Cell Mol Life Sci. 2004;61:1439-54
10. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-35
11. Sun Y, Li C, Shu Y. et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal. 2012;5:ra16
12. Mizushima N, Levine B, Cuervo AM. et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069-75
13. Hartleben B, Godel M, Meyer-Schwesinger C. et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084-96
14. Jiang M, Liu K, Luo J. et al. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176:1181-92
15. Pallet N, Bouvier N, Legendre C. et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. 2008;4:783-91
16. Periyasamy-Thandavan S, Jiang M, Wei Q. et al. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631-40
17. Yang C, Kaushal V, Shah SV. et al. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol. 2008;294:F777-87
18. Chien CT, Shyue SK, Lai MK. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation. 2007;84:1183-90
19. Suzuki C, Isaka Y, Takabatake Y. et al. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun. 2008;368:100-6
20. Inoue K, Kuwana H, Shimamura Y. et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin Exp Nephrol. 2010;14:112-22
21. Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int. 2009;76:604-13
22. Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889-92
23. Xu C, Chang A, Bradley K. et al. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014;85:72-81
24. Zhang Y, Liu G, Dull RO. et al. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am J Physiol: Lung Cell Mol Physiol. 2014;307:173-85
25. Chen X, Liu X, Wan X, Wu Y. et al. Ischemic preconditioning attenuates renal ischemia-reperfusion injury by inhibiting activation of IKKB and inflammatory response. Am J Nephrol. 2009;30:287-94
26. Kabeya Y, Mizushima N, Ueno T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720-28
27. Kadowaki M, Karim MR. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 2009;452:199-213
28. Meyer G, Czompa A, Reboul C, Csepanyi E, Czegledi A, Bak I, Balla G, Balla J, Tosaki A, Lekli I. The cellular autophagy markers Beclin-1 and LC3B-II are increased during reperfusion in fibrillated mouse hearts. Curr Pharm. 2013;19:6912-18
29. Uchino S, Bellomo R, Goldsmith D. The meaning of the blood urea nitrogen/creatinine ratio in acute kidney injury. Clin Kidney J. 2012;5:187-91
30. Nair AR, Masson GS, Ebenezer PJ, Del Piero F, Francis J. Role of TLR4 in lipopolysaccharide-induced acute kidney injury: protection by blueberry. Free Radic Biol. 2014;71:16-25
31. Patrick N, Cunningham PN, Dyanov HM, Park P, Wang J, Newell KA, Quigg RJ. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney 1. J Immunol. 2002;1168:5817-23
32. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651
33. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43-52
34. Livingston MJ, Dong Z. Autophagy in Acute Kidney Injury. Semin Nephrol. 2014;34:17-26
35. Howell GM, Gomez H Collage RD, Loughran P Zhang X, Escobar DA Billiar TR Zuckerbraun BS, Rosengart MR. Augmenting Autophagy to Treat Acute Kidney Injury during Endotoxemia in Mice. PLoS ONE. 2013;8:e69520
36. Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271-83
37. Bonventre JV. Primary proximal tubule injury leads to epithelial cell cycle arrest, fibrosis, vascular rarefaction, and glomerulosclerosis. Kidney Int. 2014(Suppl 4):39-44
38. Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Bio Med Res Int. 2014;2014:967826
39. Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902-13
40. Cunningham PN, Dyanov HM, Park P, Wang J, Newell KA, Quigg RJ. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol. 2002;168:5817-23
41. Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001-4
42. Nakahira K, Haspel JA, Rathinam VA. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222-30
43. Saitoh T, Fujita N, Jang MH. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264-8
44. Traenckner EB, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. EMBO J. 1994;13:5433-41
45. Colleran A, Ryan A, O'Gorman A. et al. Autophagosomal IkappaB alpha degradation plays a role in the long term control of tumor necrosis factor-alpha induced nuclear factor-kappaB (NF-kappaB) activity. J Biol Chem. 2011;286:22886-93
46. Criollo A, Chereau F, Malik SA. et al. Autophagy is required for the activation of NFkappaB. Cell Cycle. 2012;11:194-9
47. Mizushima N. Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728-41
48. Ravikumar B, Berger Z, Vacher C. et al. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15:1209-16
49. Behrends C, Sowa ME, Gygi SP. et al. Network organization of the human autophagy system. Nature. 2010;466:68-76
50. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-35
51. Nivon M, Richet E, Codogno P. et al. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy. 2009;5:766-83
52. Takahashi K, Mizukami H, Kamata K, Inaba W, Kato N, Hibi C, Yagihashi S. Amelioration of acute kidney injury in lipopolysaccharide-induced systemic inflammatory response syndrome by an aldose reductase inhibitor, fidarestat. PLoS One. 2012;7:e30134
53. Howell GM, Gomez H, Collage RD, Loughran P, Zhang X. et al. Augmenting Autophagy to Treat Acute Kidney Injury during Endotoxemia in Mice. PLoS One. 2013;8:e69520
54. Xu D, Chen M, Ren X, Ren X, Wu Y. Leonurine ameliorates LPS-induced acute kidney injury via suppressing ROS-mediated NF-κB signaling pathway. Fitoterapia. 2014;97:148-55
55. Zhang D, Li Y, Liu Y, Xiang X, Dong Z. Paclitaxel ameliorates LPS-induced kidney injury by binding MD-2 to block TLR4-mediated NF-κB activation and cytokine production. J Pharmacol Exp Ther. 2013;345:69-75
56. Hussain SN, Mofarrahi M, Sigala I. et al. Mechanical ventilation-induced diaphragm disuse in humans triggers autophagy. Am J Respir Crit Care Med. 2010;182:1377-86
57. Lee SJ, Smith A, Guo L. et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2011;183:649-58
58. Derde S, Vanhorebeek I, Guiza F. et al. Early parenteral nutrition evokes a phenotype of autophagy deficiency in liver and skeletal muscle of critically ill rabbits. Endocrinology. 2012;153:2267-76
59. Magnani M, Crinelli R, Bianchi M, Antonelli A. The ubiquitin-dependent proteolytic system and other potential targets for the modulation of nuclear factor-kB (NF-kB). Curr Drug Targets. 2000;1:387-99
60. Chen X, Liu X, Wan X, Wu Y, Chen Y, Cao C. Ischemic preconditioning attenuates renal ischemia-reperfusion injury by inhibiting activation of IKKbeta and inflammatory response. Am J Nephrol. 2009;30:287-94
Author contact
![]() Corresponding author: Wenhu Liu, Department of Nephrology, Beijing Friendship Hospital, Capital Medical University, No. 95 Yong An Road, Xi Cheng District, Beijing 100050, China; Email: wenhuliuccmu.edu.cn
Corresponding author: Wenhu Liu, Department of Nephrology, Beijing Friendship Hospital, Capital Medical University, No. 95 Yong An Road, Xi Cheng District, Beijing 100050, China; Email: wenhuliuccmu.edu.cn
Received 2015-4-20
Accepted 2015-7-14
Published 2015-8-1