Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(7):576-582. doi:10.7150/ijms.12111 This issue Cite
Research Paper
Serum UCH-L1 as a Novel Biomarker to Predict Neuronal Apoptosis Following Deep Hypothermic Circulatory Arrest
Ya-Ping Zhang1, Yao-Bin Zhu1 ![]() , Dayue Darrel Duan2, Xiang-Ming Fan1, Yan He1, Jun-Wu Su1, Ying-Long Liu1
, Dayue Darrel Duan2, Xiang-Ming Fan1, Yan He1, Jun-Wu Su1, Ying-Long Liu1 ![]()
1. Pediatric Heart Center, Beijing Anzhen Hospital, Capital Medical University, Beijing, China 100029
2. Laboratory of Cardiovascular Phenomics, the Department of Pharmacology, University of Nevada School of Medicine, Reno, Nevada, USA 89557
Received 2015-3-12; Accepted 2015-6-9; Published 2015-7-3
Abstract
Background: Deep hypothermic circulatory arrest (DHCA) has been used in cardiac surgery involving infant complex congenital heart disease and aortic dissection. DHCA carries a risk of neuronal apoptotic death in brain. Serum ubiquitin C-terminal hydrolase L1 (UCH-L1) level is elevated in a number of neurological diseases involving neuron injury and death. We studied the hypothesis that UCH-L1 may be a potential biomarker for DHCA-induced ischemic neuronal apoptosis.
Methods: Anesthetized piglets were used to perform cardiopulmonary bypass (CPB). DHCA was induced for 1 hour followed by CPB rewarming. Blood samples were collected and serum UCH-L1 levels were measured. Neuron apoptosis and Bax and Bcl-2 proteins in hippocampus were examined. The relationship between neuron apoptosis and UCH-L1 level was determined by receiver operating characteristics (ROC) curves and correlation analysis.
Results: DHCA resulted in marked neuronal apoptosis, significant increase in Bax:Bcl-2 ratio in hippocampus and UCH-L1 level elevations in serum (all P<0.05). Positive correlation was obtained between serum UCH-L1 level and the severity of neuron apoptosis (r= 0.78, P<0.01). By ROC, the area under the curve were 0.88 (95% CI: 0.74-0.99; P<0.01), 0.81 (95% CI: 0.81-0.96; P<0.05), 0.71 (95% CI: 0.47-0.92; P=0.11) for UCH-L1, Bax/Bcl-2 ratio and Bax, respectively. Using a cut-off point of 0.25, the UCH-L1 predicted neuronal apoptosis with a sensitivity of 85% and specificity of 57%.
Conclusion: Serum UCH-L1, as an easy and quick measurable biomarker, can predict neural apoptosis induced by DHCA. The elevation in UCH-L1 concentration is consistent with the severity of neural apoptosis following DHCA.
Keywords: ubiquitin C-terminal hydrolase-L1, neuronal apoptosis, deep hypothermic circulatory arrest, cardiopulmonary bypass
Introduction
As survival for neonates with congenital heart disease has improved, concern has shifted to the neurologic morbidity afflicting 5-40% of survivors1-4. Global cerebral ischemia, which is related to cardiopulmonary bypass (CPB) and deep hypothermic circulatory arrest (DHCA) used in the repair of cardiovascular defects, appears to play an important role in the neurologic morbidity5, 6. After a global ischemic event, many neuronal cells die by a process called apoptosis7, 8. Currently, neuronal apoptosis is difficult to identify and diagnose for clinical patients. Owing to the limitations in sensitivity and specificity, there is none of prognostic method emerging as a widely used diagnostic clinical tool for irreversible neuron apoptosis. Ubiquitin C-terminal hydrolase L1 (UCH-L1) which comprises 1-5% of total neuronal proteins is a highly abundant neuronal deubiquitinase, a cysteine protease that cleaves small peptide adducts from the C-terminus of ubiquitin9. UCH-L1 concentration has been reported to be elevated in a number of neurological diseases including aneurysmal subarachnoid hemorrhage, traumatic brain injury, stroke and neonatal hypoxic-ischemic encephalopathy10-13. It is unknown that whether the alteration in serum UCH-L1 level could potentially facilitate assessment of neuronal apoptosis following DHCA. Therefore we hypothesized that serum UCH-L1 was a novel useful biomarker to predict neuronal apoptosis following DHCA. In the present study we sought to examine the change in serum UCH-L1 level and then evaluate the relation of UCH-L1 into neuronal apoptosis in newborn piglets with DHCA.
Material and Methods
All procedures were carried out according to a protocol approved by the animal care and use committee of Anzhen Hospital, Capital Medical University. Animals were maintained and received care in Laboratory Animal Care Center of Anzhen Hospital.
Experimental Design
Nineteen piglets aged 5.43±0.53 weeks were obtained from Ke-Xing experimental animal breeding center (Beijing, China) and then were divided into 3 groups: 1) sham control, 2) CPB group and 3) DHCA group. Sham control underwent surgical preparation (n=5). CPB group underwent surgical preparation and then hypothermic CPB (n=7). DHCA group underwent surgical preparation and then hypothermic CPB and DHCA (n=7). Blood samples were collected before surgical preparation, at 6 hour and 12 hour after surgery (T1, T2 and T3, respectively). All the piglets were killed by venous injection of 15% KCl (5 ml) after 12-hour survival and then hippocampus tissues were dissected quickly.
CPB and DHCA
Firstly, surgical preparation was performed. Anesthesia was induced with intravenous ketamine (33 mg/kg) and acepromazine (3.3 mg/kg), followed by tracheal intubation, mechanical ventilation, and intravenous catheter insertion. Anesthesia was maintained with intravenous fentanyl (10ug/kg/h) and vecuronium bromide (0.4 mg/kg/h). A femoral arterial catheter was inserted to monitor arterial pressure (M1567A Transducer, Philips, Netherland). Meanwhile, electrocardiography and oxygen saturation were monitor (Servo-i, Maquet, Germany). Blood gases were determined during the whole surgery (i-STAT; Yueda Company, China). Thermistors were inserted into the cranial epidural space and rectum to monitor brain and core body temperatures (Yellow Springs, USA).
Through a right-neck incision, the carotid artery and external jugular vein were exposed, through which cannulae (Medtronic, USA) were advanced to the ascending aorta and right atrium for CPB. Heparin (400 IU/kg) was administered intravenously. After arterial cannulation, 10 ml/kg blood was collected for transfusion after CPB. The CPB circuit used a TerumoBaby RX oxygenator (Terumo, Japan) with an arterial filter (Ningbo Fly, China) receiving oxygen at a rate of 1 l/min and a nonpulsatile roller pump (Renal Systems, USA) flowing at 80 ml/kg/min. The CPB prime contained pig whole blood (100 ml), heparin (2,000 U), pancuronium (1 mg), calcium chloride (500 mg), dexamethasone (5 mg), cefazolin (25 mg/kg), furosemide (1 mg/kg), 10% calcium gluconate (10 ml) and 5% sodium bicarbonate (20 ml). Hypothermia was induced with CPB using ice bags around the head and body. At a brain temperature of 20°C (measured in the cranial epidural space), CPB was maintained for 1 hour and then rewarming was initiated in the CPB group, whereas DHCA was induced for 1 hour in the DHCA group. To rewarm, CPB pump flow and arterial perfusate were increased gradually to 100 ml/kg/h and 38°C, respectively (approximately 40 min of CPB rewarming). After 10 min of CPB, the heart was defibrillated as necessary and mechanical ventilation was resumed. CPB was discontinued when all body temperatures were greater than 32°C.
After CPB, cannulae were removed, protamine (4 mg/kg) was administered intravenously, and the neck incision was sutured closed. The blood collected before CPB was transfused over 1-2 hour to maintain a mean arterial pressure more than 50 mmHg.
Histological examination
Hippocampus tissues were removed and washed in PBS buffer. The samples were cut coronally into 2.5 mm blocks and immersed in 10% formaldehyde and then prepared for hematoxylin-eosin (HE) staining. The blocks were dehydrated in ethanol and xylene and then embedded in paraffin. One 8 μm section was cut from a paraffin block and stained with hematoxylin and eosin to determine cell damage. HE-stained slides were evaluated for neuronal cell death (photomicrograph, 400×magnification). The slides were scored semi-quantitatively on a scale of 0-5: 0 = no damage (normal neuronal structure); 1 = rare damage (<1% of neurons dead; no inflammation or infarction); 2 = mild (1-5% of neurons dead; no inflammation or infarction); 3 = moderate (6-15% of neurons dead; no inflammation or infarction); 4 = severe (16-30% of neurons dead, inflammation, or infarction); and 5 = very severe (>30% of all neurons dead; inflammation and infarction).
TUNEL assay was performed to evaluate neuron apoptosis (Meixuan Biolodical, China). After conventional dewaxing, section sample was rinsed and TUNEL reaction mixture was added. Section sample was incubated at 37℃ for 60 minutes and then washed. Agent POD was added. Section sample was incubated at 37℃ for 30 minutes and then washed. Add DAB substrate solution at room temperature and incubate for 10 minutes. Apoptosis and immunohistochemistry image analysis were performance using Axioplan 2 imaging system (ZEISS, Germany). Then apoptotic neurons were quantified. Six sections were selected for each sample and five non-duplicate HPFs (400×magnification) were selected for each section. The number of TUNEL-staining positive cells in each HPF was counted and the average of the number of apoptotic cells in each section was used.
Western Blotting
Frozen hippocampus tissue was homogenized in lysis buffer containing protease inhibitor cocktail purchased from Sigma (50 mM Tris-HCl, PH=7.4, 150 mM NaCl, 1 mM EDTA, 10 mM DTT, 0.1% SDS, 1% NP-40). The lysates were incubated on ice for 30 min and then followed by a centrifugation step at 17,000 g for 15 min at 4°C. The supernatants were collected and subjected to the Bradford Assay for protein concentration quantification (Thermo Scientific). Proteins were then added to modified Laemmli sample buffer contained DTT (150 mM final concentration) instead of 2-mercaptoethanol and denaturated at 95°C for 5 min. We loaded 20 μg protein into each lane on 10% precast Mini-Protean® TGXTM gradient gels (Bio-Rad). Proteins were transferred on to nitrocellulose membrane in 20% methanol transfer buffer with Criterion Blotter for 1 h at 100 volts. Membranes were blocked in 5% non-fat milk in PBS-T for 30 min. Bax antibody (1:200, Santa Cruz), Bcl-2 antibody (1:200, Santa Cruz), β-Actin antibody (1:500, Santa Cruz) were used for immunoblots. Secondary IgG horseradish peroxidase-linked whole antibodies from Donkey (1:5000, GE Healthcare) were used. Detection of protein was performed using AmershamTM ECLTM Prime Western Blotting Detection Reagent (GE Healthcare). Bands were visualized with ChemiDocTM MP Imaging System and quantified with quantity One software (Bio-Rad).
UCH-L1 Detection
Blood samples were collected from each piglet and then centrifuged for 10 minutes at 4000 rpm. Blood serums were immediately frozen and stored at -80℃ until the time of analysis. All blood serum samples were analyzed in duplicate and measured using a standard UCH-L1 sandwich ELISA (Rapid Bio Lab, USA) protocol as described previously14. Blank control was set up using distilled water and UCH-L1 standard curve was made (0.05 - 50 ng/well). UCH-L1 values of serum samples were calculated by comparison with standard curve.
Statistical Analysis
All data were expressed as mean±SEM. One-way ANOVA was used to analyze the data among three groups and then bonferroni correction was performed for multiple comparisons. Independent t-test was performed for comparison between two groups. The receiver operating characteristics (ROC) curves explored the relationship between neuronal apoptosis and UCH-L1. Difference was considered statistically significant when P < 0.05. All statistical tests were performed using SPSS software package 16.0 for Windows (Chicago, IL).
Results
Physiologic data
There were no significant differences in body weight among three groups (10.4±0.7 kg, 9.9±0.6 kg and 10.9±0.8 kg, P>0.05). CPB cooling and rewarming durations were similar between CPB and DHCA groups (29±8 min vs 28±9 min, 42±11 min vs 41±10, all P>0.05). The whole surgical durations for CPB and DHCA groups were 147.6±11.5 min and 154±14.2 min, respectively (P>0.05).
Neuronal apoptosis
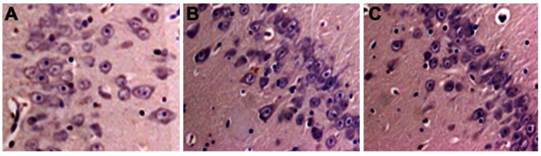
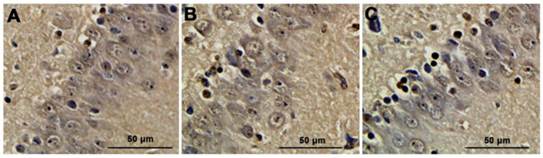
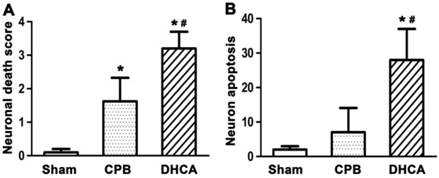
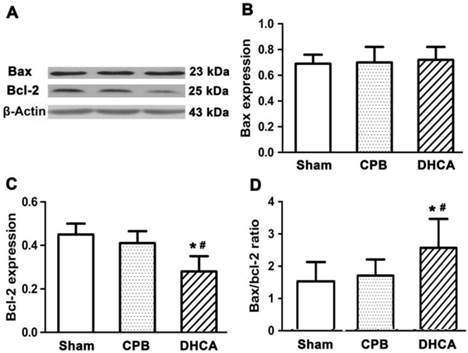
Neuronal cell death in DHCA group was severer than CPB and sham control groups (Figure 1). TUNEL assay showed that neuronal apoptosis was apparent in DHCA group whereas neuronal apoptosis was not observed in sham control group and rarely in CPB group (Figure 2). Neuronal death scores were significantly increased in both CPB and DHCA groups compared to sham operated control (Figure 3A). Furthermore, the neuronal death was more pronounced in DHCA compared to CPB alone (3.2±0.5 vs 1.6±0.7, P<0.05, Figure 3A). The number of apoptotic neurons in DHCA group was much greater than CPB and sham control groups (28±9 vs 18±7 and 2±1, all P<0.05, Figure 3B). The Bax protein expression did not change among three groups (P>0.05, Figure 4). However, Bcl-2 expression was significantly decreased in DHCA group (all P<0.05, DHCA vs CPB and Sham control, Figure 4). Hence, Bax:Bcl-2 ratio was increased in DHCA group (all P<0.05, DHCA vs CPB and Sham control, Figure 4).
Neuronal death in hippocampus. A. sham control group; B. CPB group; C. DHCA group. Necrotic cells were identified by a pyknotic nucleus or no nucleus, along with a swollen, eosinophilic cytoplasm whereas apoptotic cells were defined by the presence of nuclear karyorrhexis and minimal cytoplasmic change.
Neuronal apoptosis detected by TUNEL analysis. A. sham group; B. CPB group; C. DHCA group. Neurons containing brown yellow granules in nuclei were considered as positive apoptotic cells.
A. neuronal death score. Neuronal damage score ranged from 0 to 5, representing normal structure to severe damage, as observed on HE-stained slides. B. apoptotic neuron quantification. Values are mean ±SEM. *P < 0.05 vs sham group, #P < 0.05 vs CPB group. n=5 for sham control, n=7 for CPB and DHCA groups.
Bax and Bcl-2 protein expression by immunoblot analysis. β-Actin was used as endogenous control and calibrator. Values are mean ±SEM. *P < 0.05 vs sham control, #P < 0.05 vs CPB group. n=5 for sham control, n=7 for CPB and DHCA groups.
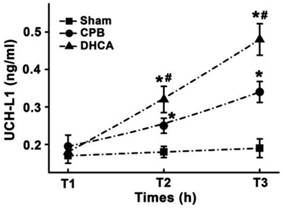
UCH-L1 level
There was no significant difference in UCH-L1 levels at T1 among three groups (Figure 5). DHCA group had highest serum UCH-L1 level and sham group exhibited lowest serum UCH-L1 concentration at both T2 (0.32±0.04, 0.25±0.03 and 0.18±0.03 for DHCA, CPB and Sham control respectively, all P<0.05, Figure 5) and T3 (0.48±0.04, 0.34±0.04, and 0.19±0.03 for DHCA, CPB and Sham control respectively, all P<0.05, Figure 5).
Serum UCH-L1 level. T1, T2 and T3 represented the time points before surgical preparation, 6 hour and 12hour after surgery, respectively. Values are mean ±SEM. *P < 0.05 vs sham control, #P < 0.05 vs CPB group. n=5 for sham control, n=7 for CPB and DHCA groups.
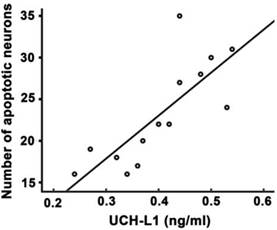
Relation of UCH-L1 into neuronal apoptosis
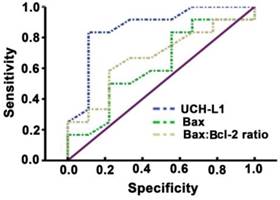
Significant positive correlation was observed between UCH-L1 level and the number of apoptotic neurons (r= 0.78, P<0.01, Figure 6). The ROC was used to explore the relationship of neuronal apoptosis with UCH-L1, Bax and Bax/Bcl-2 ratio. The area under the curve were 0.88 (95% CI: 0.74-0.99; P<0.01), 0.71 (95% CI: 0.47-0.92; P=0.11) and 0.81 (95% CI: 0.81-0.96; P<0.05), respectively (Figure 7). Using a cut-off point of 0.25, the UCH-L1 predicted neuronal apoptosis with a sensitivity of 85% and specificity of 57%.
Positive correlation between serum UCH-L1 level and the severity of neuronal apoptosis. Pearson correlation analysis was performed. r= 0.78, P<0.01.
Sensitivity and specificity of prediction of UCH-L1 for neuron apoptosis. ROC analysis was carried out. The AUC were 0.88, 0.71, 0.81 for UCH-L1, Bax and Bax:Bcl-2 ratio, respectively.
Discussion
Our findings reported the morphologic and histologic outcomes similar to those in prior investigations7, 8 but yielded evidence for UCH-L1 to predict neuronal apoptosis following DHCA. Our study provided the evidence of apoptosis after DHCA, including the presence of damaged neurons with apoptotic structure and TUNEL positivity, as well as the expression of pro-apoptotic factor Bax and anti-apoptotic factor Bcl-2. More importantly, this study demonstrated serum UCH-L1 level, as an easy and quick biochemical index, can predict DHCA-induced neuronal apoptosis and the elevation in UCH-L1 concentration is consistent with the severity of neural apoptosis following DHCA.
DHCA has been used as an adjunctive therapy with CPB to facilitate repair of complex congenital heart defects since 1970s. This technique provides a bloodless and uncluttered field for surgeons. With increased survival rates after the complex cardiac operations, obvious neurodevelopmental sequelae have been observed during long-term follow-up15-18, suggesting hypothermia-induced neurologic protection was incomplete during DHCA. After a global ischemic event induced by DHCA, neurons in the neocortex and hippocampus are selectively vulnerable to death. Previous findings with a newborn pig DHCA model suggest that cell death occurred as early as 6 hour after DHCA with a peak at 24-72 hour and many neurons in die by apoptosis 7. Apoptotic cascade was initiated as soon as 1 hour after reperfusion and continued for 72 hour after DHCA7. Neuron death and the apoptotic cascade both diminished by 1 week after DHCA7. Our model simulated neonatal heart surgery with CPB and DHCA. CPB was used to induce hypothermia and support circulation. We found that neurons in hippocampus were selectively vulnerable to die 12 hour after DHCA, many of which were apoptotic. However, cell apoptosis were not observed in hippocampus in the sham control group and were rarely founded in the CPB group.
This study showed that Bcl-2 expression was decreased and the Bax:Bcl-2 ratio was increased after DHCA. The Bcl-2 protein functions to prevent apoptosis whereas Bax protein functions to promote apoptosis19-23. The ratio of Bax:Bcl-2 dictates whether a cell will respond to proximal apoptotic stimulus. The function of Bcl-2 as anti-apoptotic protein has been explored to be via inhibition of the apoptotic cascade at a level above the interleukin β converting enzyme (ICE) group of protease24. Bcl-2 is usually distributed at the outer membranes of the nucleus, the endoplasmic reticulum, and the mitochondria25. Therefore, Bcl-2 may exert its protective effect by maintaining the integrity of these membranes and regulating calcium influx, which could be a mediator of DNA fragmentation. In our study, DHCA may stimulate neuron apoptosis by inhibiting Bcl-2 protein expression.
UCH-L1 is a neuron-specific enzyme and exists in two forms, a soluble cytoplasmic form (UCH-L1C) and a membrane-associated form (UCH-L1M). Deletion of the four C-terminal residues caused the loss of protein solubility, abrogation of substrate binding, increased cell death and an abnormal intracellular distribution. UCH-L1 has been identified previously as an index for neuronal cell injury under different neurological conditions12, 13, 26, 27. UCH-L1 elevates at a very early time point, usually within 12 hours after neuronal damage. Therefore, potential neuro-protective strategies might be very effective14. Some studies support that UCH-L1 expression regulates neuronal apoptosis28, 29. It has been reported that UCH-L1 is detectable in human serum following acute brain injuries induced by cardiac arrest30, 31. Cerebral ischemia disturbs functional and structural blood brain barrier (BBB) integrity and damaged neuron may release UCH-L1 into circulation via injured BBB after global cerebral ischemia32-35. The focus of the current study was to evaluate the relation of serum UCH-L1 level into neuronal apoptosis following DHCA. Remarkably, the data showed that DHCA group had highest serum UCH-L1 level and CPB group had higher serum UCH-L1 level than sham control within 12 hours after surgery. Significant positive correlation was obtained between serum UCH-L1 level and the severity of neuron apoptosis. The prediction of serum UCH-L1 level for neuron apoptosis at early time point had high sensitivity and mild specificity by ROC analysis.
Serum UCH-L1, an easy and quick measurable biomarker, had high sensitivity and mild specificity for the prediction of DHCA-induced neuronal apoptosis, which may be combined with neuro-radiologic methods to improve the diagnosis, prognosis and experimental therapeutic evaluation. Moreover, an elevation in UCH-L1 concentration is consistent with the severity of occurrence of apoptotic neuron death after DHCA. Future research should be directed at validation of serum UCH-L1 biomarker in large, well-designed studies.
Acknowledgements
This study is supported by the National Natural Science Foundation of China (No. 81371443 and No.81400305), Beijing Natural Science Foundation (No.7152045, No.7122056, No.7142049 and No.7142137), Beijing Talents Fund (No.2014000021469G233), Beijing Outstanding Talents Cultivation Program (No.2014000021469G233) and Beijing Municipal Health Bureau High-Level Talent Cultivation (No.2014-3-043). We thank Yao Yang for their suggestions and assistance.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Selnes OA, McKhann GM. Neurocognitive complications after coronary artery bypass surgery. Ann Neurol. 2005;57(5):615-621
2. Marino BS, Lipkin PH, Newburger JW. et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126(9):1143-1172
3. Islam MT, Hussain MZ, Bhuiyan MR, Roy GR, Barua C, Kabir A. Neurological status of children with congenital heart defects. Mymensingh Med J. 2014;23(3):538-543
4. Polito A, Barrett CS, Rycus PT, Favia I, Cogo PE, Thiagarajan RR. Neurologic Injury in Neonates with Congenital Heart Disease During Extracorporeal Membrane Oxygenation: An Analysis of ELSO Registry Data. ASAIO J. 2015;61(1):43-48
5. Liddicoat JR, Redmond JM, Vassileva CM, Baumgartner WA, Cameron DE. Hypothermic circulatory arrest in octogenarians: risk of stroke and mortality. Ann Thorac Surg. 2000;69(4):1048-1051
6. Schlunt ML, Brauer SD. Anesthetic management for the pediatric patient undergoing deep hypothermic circulatory arrest. Semin Cardiothorac Vasc Anesth. 2007;11(1):16-22
7. Ditsworth D, Priestley MA, Loepke AW. et al. Apoptotic neuronal death following deep hypothermic circulatory arrest in piglets. Anesthesiology. 2003;98(5):1119-1127
8. Kin H, Ishibashi K, Nitatori T, Kawazoe K. Hippocampal neuronal death following deep hypothermic circulatory arrest in dogs: involvement of apoptosis. Cardiovasc Surg. 1999;7(5):558-564
9. Bishop P, Rubin P, Thomson AR, Rocca D, Henley JM. UCH-L1 C-terminus plays a key role in protein stability but its farnesylation is not required for membrane association in primary neurons. J Biol Chem. 2014;289(52):36140-36149
10. Daoud H, Alharfi I, Alhelali I, Charyk ST, Qasem H, Fraser DD. Brain injury biomarkers as outcome predictors in pediatric severe traumatic brain injury. Neurocrit Care. 2014;20(3):427-435
11. Kovesdi E, Luckl J, Bukovics P. et al. Update on protein biomarkers in traumatic brain injury with emphasis on clinical use in adults and pediatrics. Acta Neurochir (Wien). 2010;152(1):1-17
12. Lewis SB, Wolper R, Chi YY. et al. Identification and preliminary characterization of ubiquitin C terminal hydrolase 1 (UCHL1) as a biomarker of neuronal loss in aneurysmal subarachnoid hemorrhage. J Neurosci Res. 2010;88(7):1475-1484
13. Douglas-Escobar M, Yang C, Bennett J. et al. A pilot study of novel biomarkers in neonates with hypoxic-ischemic encephalopathy. Pediatr Res. 2010;68(6):531-536
14. Mondello S, Palmio J, Streeter J, Hayes RL, Peltola J, Jeromin A. Ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) is increased in cerebrospinal fluid and plasma of patients after epileptic seizure. BMC Neurol. 2012;12:85
15. Mahle WT, Clancy RR, Moss EM, Gerdes M, Jobes DR, Wernovsky G. Neurodevelopmental outcome and lifestyle assessment in school-aged and adolescent children with hypoplastic left heart syndrome. Pediatrics. 2000;105(5):1082-1089
16. Wernovsky G. Current insights regarding neurological and developmental abnormalities in children and young adults with complex congenital cardiac disease. Cardiol Young. 2006;16(Suppl 1):92-104
17. Dent CL, Spaeth JP, Jones BV. et al. Brain magnetic resonance imaging abnormalities after the Norwood procedure using regional cerebral perfusion. J Thorac Cardiovasc Surg. 2006;131(1):190-197
18. Miller SP, McQuillen PS, Hamrick S. et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med. 2007;357(19):1928-1938
19. Reimers K, Choi CY, Bucan V, Vogt PM. The Bax Inhibitor-1 (BI-1) family in apoptosis and tumorigenesis. Curr Mol Med. 2008;8(2):148-156
20. Adams JM, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19(5):488-496
21. Brooks C, Dong Z. Regulation of mitochondrial morphological dynamics during apoptosis by Bcl-2 family proteins: a key in Bak? Cell Cycle. 2007;6(24):3043-3047
22. Labi V, Grespi F, Baumgartner F, Villunger A. Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ. 2008;15(6):977-987
23. Lao Y, Chang DC. Study of the functional role of Bcl-2 family proteins in regulating Ca(2+) signals in apoptotic cells. Biochem Soc Trans. 2007;35(Pt 5):1038-1039
24. Shimizu S, Eguchi Y, Kamiike W. et al. Bcl-2 blocks loss of mitochondrial membrane potential while ICE inhibitors act at a different step during inhibition of death induced by respiratory chain inhibitors. Oncogene. 1996;13(1):21-29
25. Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80(2):285-291
26. Brophy GM, Mondello S, Papa L. et al. Biokinetic analysis of ubiquitin C-terminal hydrolase-L1 (UCH-L1) in severe traumatic brain injury patient biofluids. J Neurotrauma. 2011;28(6):861-870
27. Mondello S, Papa L, Buki A. et al. Neuronal and glial markers are differently associated with computed tomography findings and outcome in patients with severe traumatic brain injury: a case control study. Crit Care. 2011;15(3):R156
28. Harada T, Harada C, Wang YL. et al. Role of ubiquitin carboxy terminal hydrolase-L1 in neural cell apoptosis induced by ischemic retinal injury in vivo. Am J Pathol. 2004;164(1):59-64
29. Reddy SS, Shruthi K, Reddy VS. et al. Altered ubiquitin-proteasome system leads to neuronal cell death in a spontaneous obese rat model. Biochim Biophys Acta. 2014;1840(9):2924-2934
30. Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16(1):1-13
31. del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thromb Res. 2000;98(3):73-81
32. Carone D, Librizzi L, Cattalini A. et al. Pravastatin acute neuroprotective effects depend on Blood brain barrier integrity in experimental cerebral ischemia. Brain Res. 2015 [Epub ahead of print]
33. Lin M, Sun W, Gong W, Zhou Z, Ding Y, Hou Q. Methylophiopogonanone A Protects against Cerebral Ischemia/Reperfusion Injury and Attenuates Blood-Brain Barrier Disruption In Vitro. PLoS One. 2015;10(4):e0124558
34. Engelhardt S, Huang SF, Patkar S, Gassmann M, Ogunshola OO. Differential responses of blood-brain barrier associated cells to hypoxia and ischemia: a comparative study. Fluids Barriers CNS. 2015;12(1):4
35. Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation. 2015;12(1):26
Author contact
![]() Corresponding authors: Prof. Ying-Long Liu, MD or Dr. Yao-Bin Zhu, MD, PhD, Pediatric Heart Center, Beijing Anzhen Hospital, Capital Medical University, 2 Anzhen Road, Beijing 100029, People's Republic of China. Tel: +86-10-64456534; Fax: +86-10-64456534; E-mail: zhuyaobinnet
Corresponding authors: Prof. Ying-Long Liu, MD or Dr. Yao-Bin Zhu, MD, PhD, Pediatric Heart Center, Beijing Anzhen Hospital, Capital Medical University, 2 Anzhen Road, Beijing 100029, People's Republic of China. Tel: +86-10-64456534; Fax: +86-10-64456534; E-mail: zhuyaobinnet