Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(5):432-440. doi:10.7150/ijms.11402 This issue Cite
Research Paper
Mitochondrial Dihydrolipoamide Dehydrogenase Is Upregulated in Response to Intermittent Hypoxic Preconditioning
Rongrong Li1,2, Xiaoting Luo1,3, Jinzi Wu1, Nopporn Thangthaeng4, Marianna E. Jung4, Siqun Jing1,5, Linya Li1, Dorette Z. Ellis1, Li Liu6, Zhengnian Ding2, Michael J. Forster4, Liang-Jun Yan1, ![]()
1. Department of Pharmaceutical Sciences, UNT System College of Pharmacy, University of North Texas Health Science Center, Fort Worth, TX 76107, USA
2. Department of Anethesiology, the First Affiliated Hospital of Nanjing University, Nanjing, Jiangsu province, China, 210029
3. Department of Biochemistry and Molecular Biology, Gannan Medical University, Ganzhou, Jiangxi province, China, 341000
4. Department of Pharmacology and Neurosciences, University of North Texas Health Science Center, Fort Worth, TX 76107, USA
5. College of Life Sciences and Technology, Xinjiang University, Urumqi, Xinjiang, China, 830046
6. Department of Geriatrics, the First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu Province, China 210029
Received 2014-12-20; Accepted 2015-5-13; Published 2015-5-23
Abstract
Intermittent hypoxia preconditioning (IHP) has been shown to protect neurons against ischemic stroke injury. Studying how proteins respond to IHP may identify targets that can help fight stroke. The objective of the present study was to investigate whether mitochondrial dihydrolipoamide dehydrogenase (DLDH) would respond to IHP and if so, whether such a response could be linked to neuroprotection in ischemic stroke injury. To do this, we subjected male rats to IHP for 20 days and measured the content and activity of DLDH as well as the three α-keto acid dehydrogenase complexes that contain DLDH. We also measured mitochondrial electron transport chain enzyme activities. Results show that DLDH content was indeed upregulated by IHP and this upregulation did not alter the activities of the three α-keto acid dehydrogenase complexes. Results also show that the activities of the five mitochondrial complexes (I-V) were not altered either by IHP. To investigate whether IHP-induced DLDH upregulation is linked to neuroprotection against ischemic stroke injury, we subjected both DLDH deficient mouse and DLDH transgenic mouse to stroke surgery followed by measurement of brain infarction volume. Results indicate that while mouse deficient in DLDH had exacerbated brain injury after stroke, mouse overexpressing human DLDH also showed increased brain injury after stroke. Therefore, the physiological significance of IHP-induced DLDH upregulation remains to be further investigated.
Keywords: dihydrolipoamide dehydrogenase, intermittent hypoxic preconditioning, ischemic stroke, mitochondria, neuroprotection
Introduction
Intermittent hypoxia (IH) has been linked to many age-related disorders such as hypertension [1, 2], sleep apnea [3], diabetes [4-6], and stroke [7, 8]. Interestingly, when applied purposely with the use of appropriate dosage, IH has been demonstrated to show preconditioning effects that can lead to numerous beneficial outcomes [9-11]; one of which is neuroprotection against ischemic stroke injury [12-16]. Therefore, studying how proteins respond to intermittent hypoxic preconditioning (IHP) in perspective of either protein expression or posttranslational modifications [17] has been of great interest lately as identification of such targets may help fight stroke.
Mitochondrial dihydrolipoamide dehydrogenase (DLDH) is an NAD+-dependent oxidoreductase [18]. It exists in three mitochondrial complexes including pyruvate dehydrogenase complex, α-ketoglutarate dehydrogenase complex, and branched chain amino acid dehydrogenase complex. DLDH is also a component of the glycine cleavage system [19]. Therefore, DLDH is a house-keeping protein and its knockout has been proven to be lethal [20]. Structurally, DLDH is a redox-sensitive enzyme because of its two cysteine residues at its active center [21, 22]. The protein can either exacerbate [23-30] or attenuate [31, 32] oxidative stress depending on the experimental conditions [33]. However, whether DLDH would respond to IHP has never been examined. The purpose of this study was thus to investigate whether DLDH expression changes in response to IHP and whether this change has any link to neuroprotection against brain ischemic injury.
Materials and methods
Chemicals
Dihydrolipoamide was synthesized from lipoamide using sodium borohydride as previously described [34, 35]. All PCR primers were purchased from Life Technologies (Carlsbad, CA). ε-amino-N-caproic acid was obtained from MP Biochemicals. Acrylamide/bisacrylamide, ammonium persulfate, Bradford protein assay solution, and Coomassie brilliant blue (CBB) R-250 were from Bio-Rad laboratories (Richmond, CA, USA). NADH, BSA, lipoamide, EDTA, and NBT chloride tablets were obtained from Sigma (St. Louis, MO, USA). Serva Blue G was purchased from Serva (Heidelberg, Germany). Rabbit anti-DLDH polyclonal antibodies (IgG) and goat anti-rabbit IgG conjugated with horseradish peroxidase were purchased from US Biological (Swampscott, MA, USA) and Invitrogen (San Diego, CA, USA), respectively. Hybond-C membrane and an ECL immunochemical detection kit were obtained from GE Healthcare (Piscataway, NJ, USA).
Intermittent hypoxia preconditioning (IHP) treatment
All animal protocols have been approved by the UNTHSC committee for animal research. An IHP program described by Ju et al. [36] was used in this study. Briefly, IHP was applied every morning for 20 days. Rats at age of 8-12 weeks were hypoxia- or sham-conditioned in 267-liter acrylic chambers that were custom-made. The IHP program consisted of brief (5-10 min) hypoxic exposures (5-8 bouts/day) with intervening 4-min reoxygenation periods [36]. Fractional inspired O2 (FIO2) in the chamber was monitored with a precision O2 sensor (Alpha Omega Instruments model 2000). Compressed nitrogen was introduced into the chamber to lower O2 content to the prescribed value within 90 s. Reoxygenation was achieved by opening the top of the chamber. Non-IHP groups underwent sham conditioning protocols in which compressed air instead of nitrogen was introduced to maintain FIO2 at 21%. Under these conditions, rats exhibited no distress during the IHP or sham conditioning sessions [36]. After the 20-day IHP treatment, rats were placed under normal conditions for 7 weeks, followed by sacrifice and tissue collection. Therefore, the whole IHP regimen contained 20-day IHP and 7-week normoxic exposure.
Mouse models
DLDH deficient mouse generated by Johnson et al [20] was obtained from Jackson laboratories via cryorecovery. DLDH transgenic mice overexpressing human DLDH (Fig. 4) was kindly generated on the background of FvBN mouse by Cyagen (Santa Clara, CA). Both DLDH deficient and DLDH transgenic mice were bred and maintained in our own colony in the animal facility of UNTHSC. All mice at age of 8-12 weeks were used throughout this study regardless of their genotypes.
Transient cerebral ischemia
For transient middle cerebral artery occlusion (tMCAO), an intraluminal filament model was used as previously described [37]. The internal carotid artery (ICA) was exposed, and a 3-0 monofilament nylon suture (0.22 ± 0.01 mm) purchased from Doccol Corporation (Sharon, MA) was introduced into the ICA lumen through a puncture and gently advanced to the distal internal carotid artery until proper resistance was felt. After 1 hour, the suture was withdrawn and the distal ICA was cauterized. At the end of 24 h reperfusion, the animals were sacrificed and the brains were harvested for either TTC staining or for mitochondria preparations (both as described below).
Measurement of infarct size
Brain ischemic damage was assessed by measuring the infarct size using 2,3,5-triphenyltetrazolium chloride (TTC) staining [37]. Briefly, brain slice was incubated for 30 minutes in a 2% solution of TTC in physiological saline at 37 o C, and then fixed in 10% formalin. The stained slice was digitally scanned and subsequently measured for the ischemic lesion size (AlphaEaseFC) [38]. The percentage of infarction volume over total brain volume was calculated as previously described [39].
Preparation of brain mitochondria
Mitochondria isolation from whole brain was carried out using Percoll gradient centrifugation as previously reported [40] with slight modifications [41, 42]. Brains were removed rapidly and homogenized in 15 ml of ice-cold, mitochondrial isolation buffer containing 0.32 M sucrose, 1 mM EDTA and 10 mM Tris-HCl, pH 7.1. The homogenate was centrifuged at 1,330 g for 10 min and the supernatant was saved. The pellet was resuspended in half volume (7.5 ml) of the original isolation buffer and centrifuged again under the same conditions. The two supernatants were combined and centrifuged further at 21,200 g for 10 min. The resulting pellet was resuspended in 12% Percoll solution prepared in mitochondrial isolation buffer followed by centrifugation at 6,900 g for 10 min. The obtained soft pellet was resuspended in 10 ml of the mitochondrial isolation buffer and centrifuged again at 6,900 g for 10 min. All of the mitochondrial pellets obtained after centrifugation were either used immediately or frozen at -80oC until analysis. Protein concentrations were determined by Bradford assay [43].
Measurement of enzyme activities
DLDH dehydrogenase activity was measured in the forward reaction or in the reverse reaction as previously described [41, 42]. Measurement of mitochondrial complexes I, IV and V activities was also conducted as previously described using in-gel based assays [44]. Activities for complexes II and III were measured spectrophotometrically as previously described [45, 46]. Pyruvate dehydrogenase complex activity was determined according to the method of Schwab et al [47] and α-keto glutarate dehydrogenase complex activity was measured by the method described by Brown and Perham [48]. Branched chain amino acid dehydrogenase complex activity was assessed according to the method of Marshall and Sokatch [49].
Polyacrylamide gel electrophoresis and Western blot analysis
Typically, 10% resolving gel of SDS-PAGE was performed unless otherwise indicated. One of the resulting gels was stained with Coomassie colloid blue [44], and the other gel was subjected to electrophoretic transfer to hybond-C membrane and immunoblotting [50]. Signals on the hybond-C membrane were visualized with an enhanced chemiluminescence kit. Nongradient blue native gel electrophoresis was performed as previously described [41]. All images were scanned by an EPSON PERFECTION 1670 scanner. All densitometric quantifications of gel images were also analyzed by AlphaEaseFC software.
Data analysis
Statistical analysis of data was performed using GraphPad's 2-tailed unpaired t test (GraphPad, San Diego, CA). A probability value less than 0.05 (p < 0.05) was considered statistically significant.
Results
DLDH activity is elevated following IHP
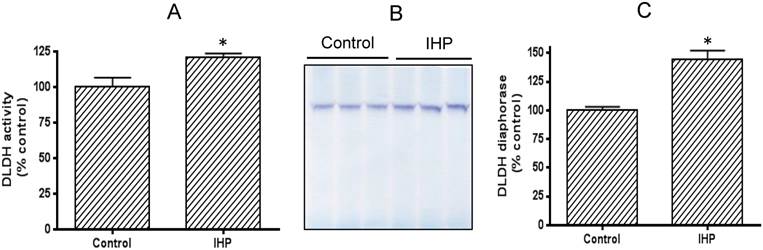
We adopted a published IHP regimen that has been shown to yield a neuroprotective effect [36]. To evaluate how DLDH expression responds to this IHP challenge, we measured DLDH activity by a spectrophotometric assay and also by blue-native gel analysis [41]. Results shown in Fig. 1A indicate that DLDH activity was significantly higher in the IHP group than in the control group. This increase was also observed on BN-PAGE (Fig. 1B) whereby densitometric quantitation also showed a significant increase (Fig. 1C).
Comparison of brain mitochondrial DLDH activities between control and IHP-treated mice. (A) Spectrophotometric assay of DLDH activity; (B) blue native gel analysis of DLDH activity; (C) densitometric quantitation of activity staining derived from (B).
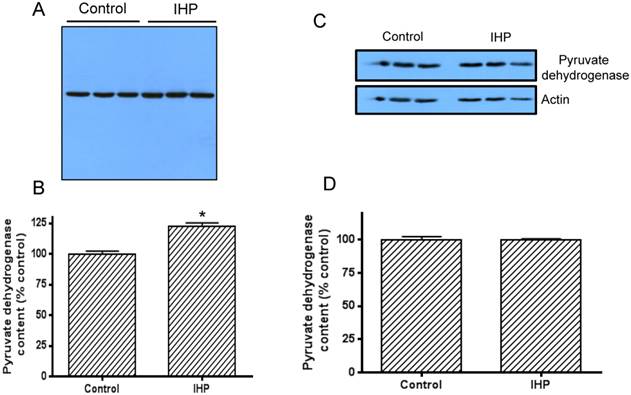
Left panel: higher DLDH protein content induced by IHP; A: Western blot assay of DLDH expression using anti-DLDH polyclonal antibodies from US Biologicals; B: densitometric quantification of the band intensity between control and IHP. Right panel, IHP did not induce a detectable change in the content of pyruvate dehydrogenase, a component of pyruvate dehydrogenase complex; C: Western blot assay of pyruvate dehydrogenase whereby actin was used as a loading control, D: densitometric quantification of the band intensity between control and IHP. N=3, *p<0.05. Note: the Western blot signal was developed on a film using X-ray developer.
Elevated DLDH activity by IHP is due to an increased DLDH protein expression
To investigate whether this IHP induced change in DLDH activity was due to the change in protein expression, we then measured DLDH protein levels by western blot assay using anti-DLDH antibodies. Results show that DLDH expression was indeed higher in the IHP group than in the control group (Fig. 2A), and a densitometric quantification indicates that this increase was significant (Fig. 2B). Further investigation indicated that this increase in DLDH protein content was not due to an increase in mitochondrial mass, as the content of mitochondrial pyruvate dehydrogenase, the first component of pyruvate dehydrogenase complex, was not increased after the IHP treatment whereby actin was used as a loading control (Fig. 2, C and D).
IHP does not alter the enzyme activities of the three α-keto acid dehydrogenase complexes that contain DLDH and the activities of the five mitochondrial complexes (I to V)
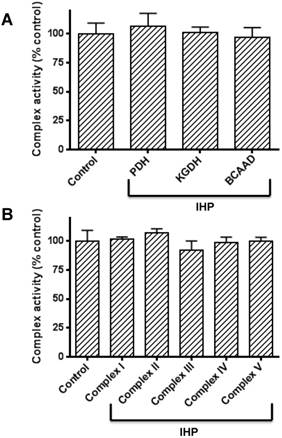
The above results clearly show that DLDH expression was upregulated by IHP. As DLDH exists in three α-keto acid dehydrogenase complexes, we wondered if the activities of the three enzyme complexes would also show changes after IHP treatment. Accordingly, we measured the enzymes' activities of all the three complexes by spectrophotometric assays. Results in Fig. 3A show that none of the complexes' activity changed in response to IHP, implicating that DLDH was upregulated independent of the other two components of the enzyme complexes whereby a fixed ratio exists between each of the three components [51]. Moreover, the activities of the five mitochondrial complexes (I to V) in the mitochondrial membrane involved in electron transport chain and ATP production were not altered either (Fig. 3B).
Both DLDH deficiency and DLDH overexpression increase ischemic brain injury
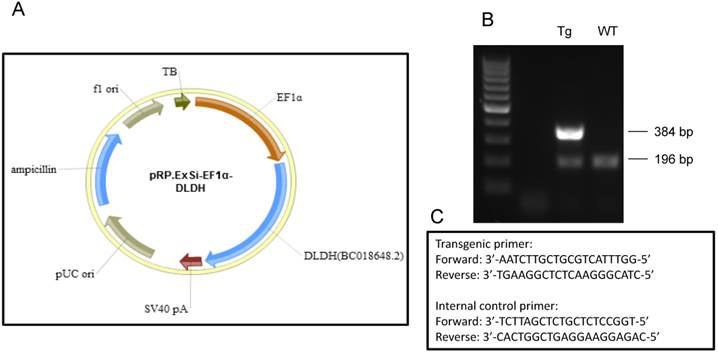
As IHP is known to induce neuroprotective effect in stroke [14, 52], we reasoned that changes in DLDH expression might be linked to neurodegeneration or neuroprotection after stroke, depending on whether DLDH is down- or up-regulated. To test these possibilities, we used two mouse models. One was DLDH deficient mouse in which only 50% DLDH is expressed when compared to wildtype mice [20], and the other was DLDH transgenic mouse in which human DLDH was globally overexpressed. As shown in Fig. 4, a transgenic vector (pRP.ExSi-EF1α-DLDH, Fig. 4A) was constructed followed by injection into the pronucleus of fertilized eggs in FvBN strain. The injected eggs were implanted into surrogate mothers afterwards. The mentioned transgenic founders and their offspring were identified by PCR genotyping (Fig. 4B) using the primers as shown in Fig. 4C, whereby the internal control primers for the wildtype mice were also given.
Measurements of activities of α-keto acid dehydrogenase complexes containing DLDH and activities of mitochondrial oxidative phosphorylation complexes I to V following IHP. (A) Activities of the three α-keto acid dehydrogenase complexes; (B) Activities of complexes I to V. Please refer to the text for detailed methods.
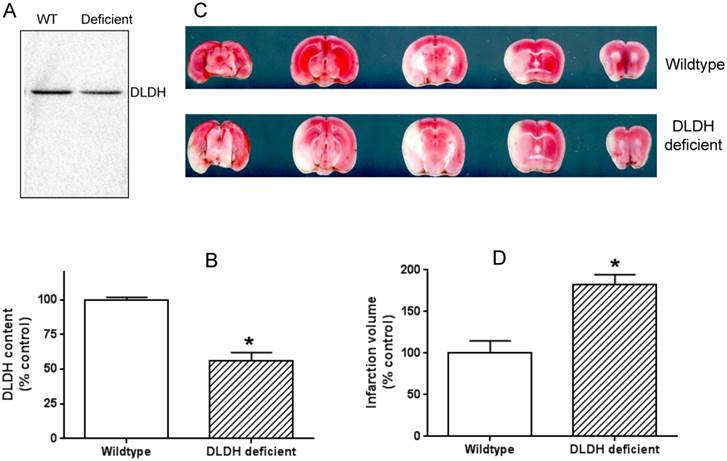
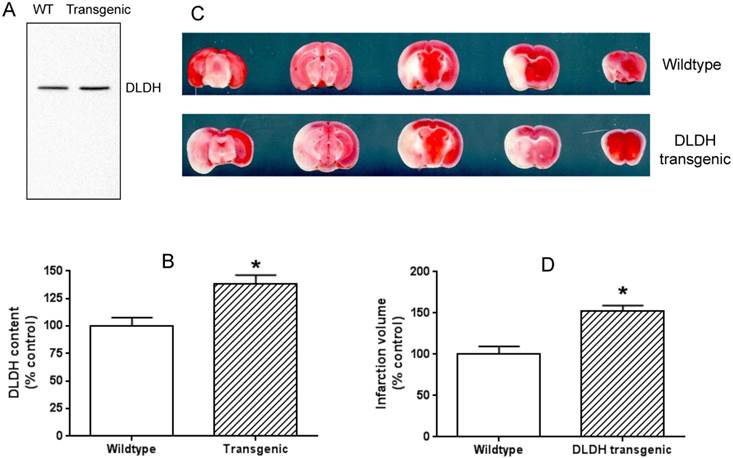
The idea of using the two mouse models was that a lower level of DLDH in the DLDH deficient mouse would render the mouse more vulnerable to stroke injury given the observation that DLDH deficient mouse is more susceptible to chemical neurotoxicity [53], while a high level of DLDH would render some degree of tolerance to stroke injury based on the establishment that excess DLDH can exist independently of the α-keto acid dehydrogenase complexes and can act as an antioxidant [31, 32]. Results indicate that DLDH content in the DLDH deficient mouse was indeed nearly 50% lower than that in the wildtype mouse (Fig. 5 A and B); and brain infarction volume in DLDH deficient mouse was indeed greater than that in the wildtype mouse (Fig. 5 C and D). Unexpectedly, brain infarction volume in the transgenic mouse was also greater than that in the control mouse (Fig. 6 C and D), though DLDH content in the transgenic mouse was higher than that in the wildtype mouse (Fig. 6 A and B). These results indicate that while DLDH deficiency exacerbated brain ischemic injury, overexpression of DLDH also worsened brain ischemic injury, at least for this transgenic mouse model under our experimental conditions.
Overexpressed human DLDH in the mouse lacks enzyme activity
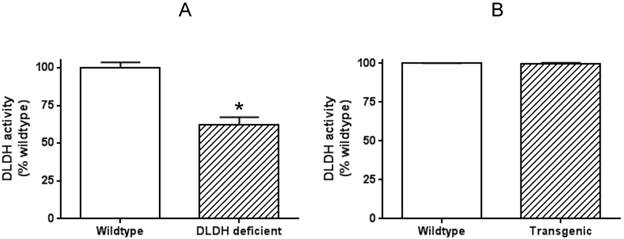
To explore why human DLDH overexpression in the mouse model did not confer neuroprotection against ischemic stroke injury, we further measured DLDH activity in the brains of both wildtype and transgenic mice, along with that in DLDH deficient mice. Results in Fig. 7 indicate that while there was a 40%-50% decrease in DLDH activity in DLDH deficient mouse, the enzyme activity in the transgenic mouse model did not show any increase when compared with that of the wildtype mouse. Such results indicate that the overexpressed mitochondrial DLDH lacked enzyme activity and might be toxic to the brain given that inactive proteins may aggregate to elicit neurotoxicity.
(A) Map of pRP.ExSi-EF1a-DLDH vector containing the human DLD gene used for generation of the transgenic mouse model that globally overexpresses human DLDH. (B) Agarose gel electrophoresis of PCR genotyping products from the transgenic and wildtype. PCR product showing the existence of only 196 base pairs is that of wildtype mouse, while that showing an additional band containing 384 base pairs is that of transgenic mouse. (C) PCR genotyping primers used for identification of the transgenic mice and the corresponding control mice following breeding.
DLDH deficiency exacerbated brain ischemic injury after ischemic stroke. (A) a lower DLDH content in the DLDH deficient mouse than that in the wildtype mouse; (B) densitometric quantification of DLDH expression derived from Western blot assays as shown in (A), N = 3, *p<0.05; (C) comparison of brain infarction volume between wildtype and DLDH deficient mice; (D) densitometric quantification of the infarction volume as shown in (C), N = 6, p<0.05. Note: the Western blot signal in this figure and that in Figure 6 was developed using a Bio-Rad digital imaging system (ChemiDocTM MP System).
Overexpression of human DLDH aggravated brain ischemic injury after ischemic stroke. (A) a higher DLDH content in the transgenic mouse than that in the wildtype mouse; (B) densitometric quantification of DLDH expression derived from Western blot assays as shown in (A), N = 3, *p<0.05; (C) comparison of brain infarction volume between wildtype and DLDH transgenic mice; (D) densitometric quantification of the infarction volume as shown in (C), N =6, p<0.05.
Measurement of brain DLDH activities in DLDH deficient and DLDH transgenic mice, respectively, along with that of corresponding wildtype mouse. Shown are (A) DLDH activities between DLDH deficient mouse and wildtype mouse. (B) DLDH activities between transgenic mouse and wildtype mouse.
Discussion
In the present study, we have demonstrated that DLDH responds to IHP by upregulating its protein content in the rat brain (Figs. 1 and 2). We further demonstrated that the mitochondrial complexes containing DLDH did not show any detectable activity changes after IHP treatment (Fig. 3A), indicating that DLDH is upregulated by the IHP regimen without changes in the activities of the three α-keto acid dehydrogenase complexes. Additionally, we have also demonstrated that the activities of the five mitochondrial complexes (I-V) did not show detectable changes either (Figs. 3B). It should be noted that DLDH upregulation by IHP was not due to any possible increase in mitochondrial mass as the protein content of pyruvate dehydrogenase was not changed by IHP (Fig. 2 C and D). Moreover, while DLDH deficiency expectedly increased brain ischemic injury (Fig. 5), DLDH overexpression also exacerbated brain ischemic injury as DLDH transgenic mouse also showed an increased infarction volume after ischemic stroke surgery (Fig. 6).
DLDH is a component of the α-keto acid dehydrogenase complexes and exists proportionally to the other two components within each complex [51, 54, 55]. For example, in eukaryotic pyruvate dehydrogenase complex, there are 30 subunits of pyruvate decarboxylase, 60 subunits of dihydrolipoyl transacetylase, and 12 subunits of DLDH. Hence overexpression of any of the three components will not be able to be incorporated into the whole complex if the other two components are not proportionally overexpressed. Instead, the overexpressed component will likely exist in excess and may freely float in mitochondria. This seems to be the case for DLDH upregulation by IHP as reflected by anti-DLDH Western blot assay of mitochondrial preparation (Fig. 1). It should be noted that it is possible that the change in DLDH activity or expression is caused by other changes taking place upstream of DLDH gene expression, which remains unknown at this time. Additionally, as we used whole brain instead of a specific brain region for mitochondria isolation and analysis, the increase in DLDH expression by IHP should truly reflect the overall DLDH response to IHP challenges.
As upregulated DLDH existed independently of the three α-keto acid dehydrogenase complexes in mitochondria, we reasoned that this extra DLDH might be functioning as an antioxidant. In other words, the function of DLDH itself other than the participation in mitochondrial keto acid dehydrogenase complexes may be neuroprotective. Accordingly, we overexpressed human DLDH in a mouse model, then performed ischemic stroke on these mice. Unexpectedly, however, the infarction volume in the brain of transgenic mice was actually greater than that in the control mice (Fig. 6C), a result similar to what was found in the DLDH deficient mouse model (Fig. 5C), which is known to be susceptible to neurotoxic challenges [53]. The reason for an increased brain injury in the transgenic mouse could be due to the observation that overexpressed human DLDH is not active (Fig. 7), which indicates that such overexpressed DLDH is not acting as an antioxidant enzyme, at least in our experimental system.
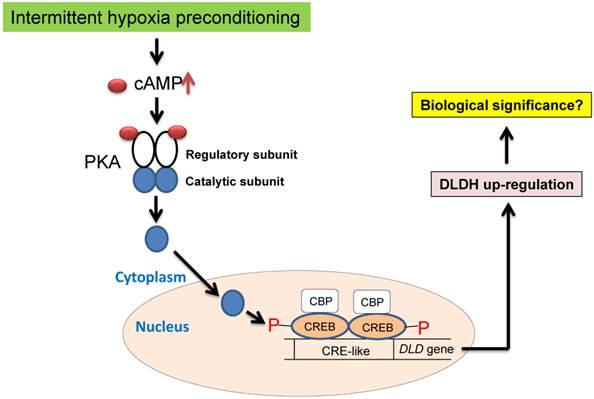
We attempted to elucidate the mechanisms of DLDH upregulation under the conditions of oxygen glucose deprivation (OGD) using SH-SY5Y neuronal cells [56]. Unfortunately, we failed to observe DLDH upregulation after the OGD treatment. This result indicates a discrepancy of DLDH response to hypoxia between live animals and cultured cells. Nonetheless, in the literature, it is well known that DLDH expression is controlled by the transcription factor cAMP response element-binding protein (CREB) [57, 58], which can be activated by IHP via cAMP activation of protein kinase A [59-61] (Fig. 8) and is known to be involved in preconditioning against stroke injury [59-61]. Future studies may need to be conducted to confirm this mechanism using animals under the IHP conditions. Additionally, there might also be a role for Ca2+ in activating the CREB signaling pathway. In fact, it is well established that increased CREB phosphorylation and increased CREB binding to CRE may also be partly regulated by the Ca2+/calmodulin-regulated protein kinase (CaMK) signaling pathway under hypoxic conditions [62-65]. Therefore, future studies may also be needed to investigate the involvement of this signaling pathway in DLDH upregulation by IHP.
In summary, we report herein that DLDH was upregulated by IHP and this upregulation occurred without changes in mitochondrial function. The α-keto acid dehydrogenase complexes containing DLDH also didn't show alterations in their enzyme activities after IHP. As both DLDH deficient mouse and DLDH transgenic mouse showed a significant increase in ischemic brain injuries, the physiological or pathophysiological significance of DLDH upregulation by IHP is unknown at this time and remains to be further explored.
Proposed signaling pathways of IHP-induced DLDH upregulation via the cAMP/CREB signaling pathway.
Acknowledgements
This work was supported in part by National Institute of Neurological Disorders and Stroke (R01NS079792 to L. J. Y.).
Conflict of interests
The authors declare that there is no conflict of interests.
References
1. Mohsenin V. Obstructive sleep apnea and hypertension: a critical review. Current hypertension reports. 2014;16:482. doi:10.1007/s11906-014-0482-4
2. Marshall JM. Development of hypertension in chronic intermittent hypoxia: is it driven by cardiac output rather than by peripheral resistance? Experimental physiology. 2014;99:1286-7 doi:10.1113/expphysiol.2014.082560
3. May AM, Mehra R. Obstructive sleep apnea: role of intermittent hypoxia and inflammation. Seminars in respiratory and critical care medicine. 2014;35:531-44 doi:10.1055/s-0034-1390023
4. Lam JC, Mak JC, Ip MS. Obesity, obstructive sleep apnoea and metabolic syndrome. Respirology. 2012;17:223-36 doi:10.1111/j.1440-1843.2011.02081.x
5. Seetho IW, Wilding JP. Sleep-disordered breathing, type 2 diabetes and the metabolic syndrome. Chronic respiratory disease. 2014 doi:10.1177/1479972314552806
6. Torrella M, Castells I, Gimenez-Perez G, Recasens A, Miquel M, Simo O. et al. Intermittent hypoxia is an independent marker of poorer glycaemic control in patients with uncontrolled type 2 diabetes. Diabetes & metabolism. 2015 doi:10.1016/j.diabet.2015.01.002
7. Bagai K. Obstructive sleep apnea, stroke, and cardiovascular diseases. The neurologist. 2010;16:329-39 doi:10.1097/NRL.0b013e3181f097cb
8. Wang Y, Guo SZ, Bonen A, Li RC, Kheirandish-Gozal L, Zhang SX. et al. Monocarboxylate transporter 2 and stroke severity in a rodent model of sleep apnea. J Neurosci. 2011;31:10241-8 doi:10.1523/JNEUROSCI.1462-11.2011
9. Ding HL, Zhu HF, Dong JW, Zhu WZ, Zhou ZN. Intermittent hypoxia protects the rat heart against ischemia/reperfusion injury by activating protein kinase C. Life Sci. 2004;75:2587-603 doi:10.1016/j.lfs.2004.07.005
10. Dale EA, Ben Mabrouk F, Mitchell GS. Unexpected benefits of intermittent hypoxia: enhanced respiratory and nonrespiratory motor function. Physiology. 2014;29:39-48 doi:10.1152/physiol.00012.2013
11. Navarrete-Opazo AA, Mitchell GS. Therapeutic Potential of Intermittent Hypoxia: A Matter of Dose. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1181-R97 doi:10.1152/ajpregu.00208.2014
12. Zhu LL, Zhao T, Li HS, Zhao H, Wu LY, Ding AS. et al. Neurogenesis in the adult rat brain after intermittent hypoxia. Brain Res. 2005;1055:1-6 doi:10.1016/j.brainres.2005.04.075
13. Leconte C, Tixier E, Freret T, Toutain J, Saulnier R, Boulouard M. et al. Delayed hypoxic postconditioning protects against cerebral ischemia in the mouse. Stroke. 2009;40:3349-55
14. Stowe AM, Altay T, Freie AB, Gidday JM. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol. 2011;69:975-85
15. Sharp FR, Ran R, Lu A, Tang Y, Strauss KI, Glass T. et al. Hypoxic preconditioning protects against ischemic brain injury. NeuroRx. 2004;1:26-35
16. Ran R, Xu H, Lu A, Bernaudin M, Sharp FR. Hypoxia preconditioning in the brain. Dev Neurosci. 2005;27:87-92
17. Yan LJ. Protein Redox Modification as a Cellular Defense Mechanism against Tissue Ischemic Injury. Oxidative Medicine and Cellular Longevity. 2014;2014:12. doi:10.1155/2014/343154
18. Williams CH Jr. Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase-a family of flavoenzyme transhydrogenases. In: (ed.) Muller F. Chemistry and Biochemistry of Flavoenzymes. Boca Raton: CRC Press. 1992:121-212
19. Kikuchi G, Hiraga K. The mitochondrial glycine cleavage system. Unique features of the glycine decarboxylation. Mol Cell Biochem. 1982;45:137-49
20. Johnson MT, Yang HS, Magnuson T, Patel MS. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci U S A. 1997;94:14512-7
21. Ciszak EM, Makal A, Hong YS, Vettaikkorumakankauv AK, Korotchkina LG, Patel MS. How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J Biol Chem. 2006;281:648-55
22. Brautigam CA, Chuang JL, Tomchick DR, Machius M, Chuang DT. Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J Mol Biol. 2005;350:543-52
23. Bando Y, Aki K. Mechanisms of generation of oxygen radicals and reductive mobilization of ferritin iron by lipoamide dehydrogenase. J Biochem (Tokyo). 1991;109:450-4
24. Sreider CM, Grinblat L, Stoppani AO. Catalysis of nitrofuran redox-cycling and superoxide anion production by heart lipoamide dehydrogenase. Biochem Pharmacol. 1990;40:1849-57
25. Gazaryan IG, Krasnikov BF, Ashby GA, Thorneley RN, Kristal BS, Brown AM. Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J Biol Chem. 2002;277:10064-72
26. Tahara EB, Barros MH, Oliveira GA, Netto LE, Kowaltowski AJ. Dihydrolipoyl dehydrogenase as a source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomyces cerevisiae aging. Faseb J. 2007;21:274-83
27. Ambrus A, Torocsik B, Tretter L, Ozohanics O, Adam-Vizi V. Stimulation of reactive oxygen species generation by disease-causing mutations of lipoamide dehydrogenase. Hum Mol Genet. 2011;20:2984-95
28. Zhang Q, Zou P, Zhan H, Zhang M, Zhang L, Ge RS. et al. Dihydrolipoamide dehydrogenase and cAMP are associated with cadmium-mediated Leydig cell damage. Toxicol Lett. 2011;205:183-9
29. Kareyeva AV, Grivennikova VG, Cecchini G, Vinogradov AD. Molecular identification of the enzyme responsible for the mitochondrial NADH-supported ammonium-dependent hydrogen peroxide production. FEBS Lett. 2011;585:385-9
30. Kareyeva AV, Grivennikova VG, Vinogradov AD. Mitochondrial hydrogen peroxide production as determined by the pyridine nucleotide pool and its redox state. Biochim Biophys Acta. 2012
31. Korotchkina LG, Yang H, Tirosh O, Packer L, Patel MS. Protection by thiols of the mitochondrial complexes from 4-hydroxy-2-nonenal. Free Radic Biol Med. 2001;30:992-9
32. Igamberdiev AU, Bykova NV, Ens W, Hill RD. Dihydrolipoamide dehydrogenase from porcine heart catalyzes NADH-dependent scavenging of nitric oxide. FEBS Lett. 2004;568:146-50
33. Yan LJ, Sumien N, Thangthaeng N, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic Res. 2013;47:123-33
34. Patel MS, Vettakkorumakankav NN, Liu TC. Dihydrolipoamide dehydrogenase: activity assays. Methods Enzymol. 1995;252:186-95
35. Patel MS, Hong YS. Lipoic acid as an antioxidant: the role of dihydrolipoamide dehydrogenase. In: (ed.) Armstrong D. Free Radical and Antioxidant Protocols. Totowa, NJ: Humana Press. 1998:337-46
36. Ju X, Mallet RT, Downey HF, Metzger DB, Jung ME. Intermittent hypoxia conditioning protects mitochondrial cytochrome c oxidase of rat cerebellum from ethanol withdrawal stress. J Appl Physiol. 2012;112:1706-14
37. Yang SH, Liu R, Wen Y, Perez E, Cutright J, Brun-Zinkernagel AM. et al. Neuroendocrine mechanism for tolerance to cerebral ischemia-reperfusion injury in male rats. J Neurobiol. 2005;62:341-51
38. Ma Y, Lu C, Li C, Li R, Zhang Y, Ma H. et al. Overexpression of HSPA12B protects against cerebral ischemia/reperfusion injury via a PI3K/Akt-dependent mechanism. Biochim Biophys Acta. 2013;1832:57-66 doi:10.1016/j.bbadis.2012.10.003
39. Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL. et al. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. Journal of neuroimmunology. 2008;199:75-82 doi:10.1016/j.jneuroim.2008.05.009
40. Sims NR. Methods in Toxicology: Mitochondrial Dysfunction. San Diego: Academic Press. 1993
41. Yan LJ, Yang SH, Shu H, Prokai L, Forster MJ. Histochemical staining and quantification of dihydrolipoamide dehydrogenase diaphorase activity using blue native PAGE. Electrophoresis. 2007;28:1036-45
42. Yan LJ, Thangthaeng N, Forster MJ. Changes in dihydrolipoamide dehydrogenase expression and activity during postnatal development and aging in the rat brain. Mech Ageing Dev. 2008;129:282-90
43. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-54
44. Yan LJ, Forster MJ. Resolving mitochondrial protein complexes using nongradient blue native polyacrylamide gel electrophoresis. Anal Biochem. 2009;389:143-9
45. Tripathy MK, Mitra D. Differential modulation of mitochondrial OXPHOS system during HIV-1 induced T-cell apoptosis: up regulation of Complex-IV subunit COX-II and its possible implications. Apoptosis: an international journal on programmed cell death. 2010;15:28-40 doi:10.1007/s10495-009-0408-9
46. Gusdon AM, Votyakova TV, Reynolds IJ, Mathews CE. Nuclear and mitochondrial interaction involving mt-Nd2 leads to increased mitochondrial reactive oxygen species production. J Biol Chem. 2007;282:5171-9
47. Schwab MA, Kolker S, van den Heuvel LP, Sauer S, Wolf NI, Rating D. et al. Optimized spectrophotometric assay for the completely activated pyruvate dehydrogenase complex in fibroblasts. Clin Chem. 2005;51:151-60
48. Brown JP, Perham RN. Selective inactivation of the transacylase components of the 2-oxo acid dehydrogenase multienzyme complexes of Escherichia coli. Biochem J. 1976;155:419-27
49. Marshall VD, Sokatch JR. Regulation of valine catabolism in Pseudomonas putida. J Bacteriol. 1972;110:1073-81
50. Yan LJ, Orr WC, Sohal RS. Identification of oxidized proteins based on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, immunochemical detection, isoelectric focusing, and microsequencing. Anal Biochem. 1998;263:67-71
51. Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990;4:3224-33
52. Stowe AM, Wacker BK, Cravens PD, Perfater JL, Li MK, Hu R. et al. CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J Neuroinflammation. 2012;9:33
53. Klivenyi P, Starkov AA, Calingasan NY, Gardian G, Browne SE, Yang L. et al. Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity. J Neurochem. 2004;88:1352-60
54. Patel MS, Harris RA. Mammalian alpha-keto acid dehydrogenase complexes: gene regulation and genetic defects. Faseb J. 1995;9:1164-72
55. Zhou ZH, McCarthy DB, O'Connor CM, Reed LJ, Stoops JK. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci U S A. 2001;98:14802-7
56. Lin WY, Chang YC, Ho CJ, Huang CC. Ischemic preconditioning reduces neurovascular damage after hypoxia-ischemia via the cellular inhibitor of apoptosis 1 in neonatal brain. Stroke. 2013;44:162-9 doi:10.1161/STROKEAHA.112.677617
57. Yang HS, Morris JI, Wang Q, Korotchkina LG, Kwon M, Patel MS. Human dihydrolipoamide dehydrogenase gene transcription is mediated by cAMP-response element-like site and TACGAC direct repeat. Int J Biochem Cell Biol. 2001;33:902-13
58. Johanning GL, Morris JI, Madhusudhan KT, Samols D, Patel MS. Characterization of the transcriptional regulatory region of the human dihydrolipoamide dehydrogenase gene. Proc Natl Acad Sci U S A. 1992;89:10964-8
59. Navon H, Bromberg Y, Sperling O, Shani E. Neuroprotection by NMDA preconditioning against glutamate cytotoxicity is mediated through activation of ERK 1/2, inactivation of JNK, and by prevention of glutamate-induced CREB inactivation. J Mol Neurosci. 2012;46:100-8
60. Li XD, Cheng YT, Yang YJ, Meng XM, Zhao JL, Zhang HT. et al. PKA-mediated eNOS phosphorylation in the protection of ischemic preconditioning against no-reflow. Microvasc Res. 2012;84:44-54
61. Lin WY, Chang YC, Lee HT, Huang CC. CREB activation in the rapid, intermediate, and delayed ischemic preconditioning against hypoxic-ischemia in neonatal rat. J Neurochem. 2009;108:847-59 doi:10.1111/j.0022-3042.2008.05828.x
62. Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821-61
63. Seta KA, Yuan Y, Spicer Z, Lu G, Bedard J, Ferguson TK. et al. The role of calcium in hypoxia-induced signal transduction and gene expression. Cell Calcium. 2004;36:331-40
64. Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS. et al. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron. 2002;34:221-33
65. Goldbart A, Row BW, Kheirandish L, Schurr A, Gozal E, Guo SZ. et al. Intermittent hypoxic exposure during light phase induces changes in cAMP response element binding protein activity in the rat CA1 hippocampal region: water maze performance correlates. Neuroscience. 2003;122:585-90
Author contact
![]() Corresponding author: Department of Pharmaceutical Sciences, UNT System College of Pharmacy, University of North Texas Health Science Center, 3500 Camp Bowie Blvd, Fort Worth TX 76107. Phone 817-735-2386, Fax 817-735-2603. Email: liang-jun.yanedu
Corresponding author: Department of Pharmaceutical Sciences, UNT System College of Pharmacy, University of North Texas Health Science Center, 3500 Camp Bowie Blvd, Fort Worth TX 76107. Phone 817-735-2386, Fax 817-735-2603. Email: liang-jun.yanedu