Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(3):288-294. doi:10.7150/ijms.10527 This issue Cite
Research Paper
FDG-PET and NeuN-GFAP Immunohistochemistry of Hippocampus at Different Phases of the Pilocarpine Model of Temporal Lobe Epilepsy
Liang Zhang1, Yi Guo1, Haitao Hu1, Jing Wang2, Zhirong Liu1, Feng Gao1 ![]()
1. Department of Neurology, Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, China;
2. Key Laboratory of Medical Molecular Imaging of Zhejiang Province, Hangzhou, China.
Received 2014-9-11; Accepted 2015-2-8; Published 2015-3-19
Abstract
Purpose: Hippocampal glucose hypometabolism has been implicated in the pathogenesis of temporal lobe epilepsy (TLE). However, the underlying pathophysiological basis for this hypometabolism remains elusive. The aim of this study was to investigate the relationship between hippocampal hypometabolism and the histological changes seen in rats after systemic pilocarpine treatment.
Methods: 18F-fluorodeoxyglucose (FDG) small-animal positron emission tomography (microPET) was performed on day zero (untreated), day seven (latent) and day sixty (chronic phase) after the initial status epilepticus. The microPET imaging data were correlated with the immunoreactivity of neuron-specific nuclear protein (NeuN) and glial fibrillary acidic protein (GFAP) in the hippocampus at each time point.
Results: 18F-FDG-microPET images showed the hippocampus presented with persistent hypometabolism during epileptogenesis and partly recovered in the chronic phase. Hippocampal glucose uptake defects correlate with NeuN immunoreactivity in the latent phase and GFAP immunoreactivity in the chronic phase.
Conclusions: Severe glucose hypometabolism in the hippocampus during the latent phase correlates with neuronal cell loss. The partial recovery of hippocampal glucose uptake in the chronic phase may be due to astrogliosis.
Keywords: Temporal lobe epilepsy, Pilocarpine, Neurons, Astrocytes, Glucose uptake, microPET.
Introduction
Hippocampus glucose hypometabolism is common in patients with drug-resistant temporal lobe epilepsy (TLE). It is also often specific enough to identify the seizure onset zone [1-3]. Some studies have shown that hypometabolism may be associated with tissue degeneration and cell loss in these zones [4, 5]. However, recently a number of studies have suggested that hippocampal hypometabolism may be a consequence of neural plasticity in the epileptic brain rather than tissue damage. Furthermore, it has been suggested that it can be reversed through surgical removal of the epileptic focus [6]. To date, the underlying cause of hippocampal hypometabolism remains elusive.
It is difficult to investigate the pathophysiological basis of hypometabolism in humans' epileptogenesis as the early epileptogenic stage is not easily found in most patients. The pilocarpine model of epilepsy in rats has been of special interest because it mimics the temporal and developmental aspects of human TLE and, thus offers the advantage of allowing assessment of the baseline, developing and chronic epileptic states. Small-animal positron emission tomography (microPET) can be applied in in vivo epilepsy research to quantitatively determine the rates of various physiological and biochemical processes with minimal invasiveness [7]. As a well-known radiotracer, 18F-FDG has been frequently used to evaluate glucose metabolic activity. Our previous study using 18F-FDG- microPET revealed that specific temporospatial changes occur with regard to glucose utilization in rats, which were noted to present with persistent hippocampal hypometabolism during the course of epileptogenesis after treatment with pilocarpine [8]. However, the pathophysiological basis for the observed hypometabolism was not disclosed. A prominent neuronal loss, the most conspicuous finding in the hippocampus is the common pathology of the TLE. Additionally, astroglial activation is supposed as an important cell response during TLE [9].
In the present study, we investigate glucose utilization in rat hippocampus at different time window after pilocarpine administration using in vivo 18F-FDG-microPET. Images were collected prior to pilocarpine treatment, during the latent phase (7 days after initial status epilepicuh, SE) and during the chronic phase (60 days after initial SE). To assess the contribution of various cellular alterations that occur in parallel with glucose utilization to the microPET signal, we also investigated the immunoreactivity of neuron-specific nuclear protein (NeuN) and glial fibrillary acidic protein (GFAP) in the hippocampus at each time point.
Materials and methods
Animals
Male Sprague-Dawley rats (260-300 g, Grade II, Experimental Animal Center, Zhejiang Academy of Medical Science, Hangzhou, China) were maintained in individual cages with a 12 h/12 h light-dark cycle. Water and food were given ad libitum. All experiments were carried out in accordance with the ethical guidelines of the Animal Experimentation Committee of Zhejiang University and were in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Pilocarpine protocol
The pilocarpine protocol was the same as our previous study [8]. Briefly, 36 rats were injected with pilocarpine (360 mg kg-1, intraperitoneal injection (i.p.); P6503, Sigma, USA) to induce SE. Methscopolamine bromide (1 mg kg-1, i.p.; S8502, Sigma, USA) was injected 30 min prior to the administration of pilocarpine to reduce its peripheral effects. If the rat did not develop SE within 45 min, an additional dose of pilocarpine (110 mg.kg-1) was given. All rats received a single dose of diazepam (20 mg kg-1, i.p.; D0899, Sigma, USA) 120 min after SE onset. 24 rats treated with the same volume saline were used as control (8 in each time point). Twelve rats died after SE onset and were excluded from this study.
18F-FDG-microPET imaging
Three serial 18F-FDG-microPET images were acquired for 24 animals over a period of 60 days as follows: (1) 2 weeks before SE (baseline); (2) 7 days following SE (latent phase); and (3) 60 days following SE (chronic phase). After each imaging session, eight rats in each group were sacrificed and the brains were obtained for immunohistochemical staining.
The regional glucose uptake of the brain was measured using the same method as previously reported [8, 10, 11]. Under a brief (1.5 min) halothane gas anesthesia, the rats were intravenously injected with 500 µCi mmol-1 18F-FDG through the dorsal penile vein. Subsequently, the animals were returned to individual cages and placed in a room with minimal ambient noise. The rats were allowed to freely move for 40 min following the tracer injection in order to reduce the influence of the anesthesia on brain metabolism [12]. Subsequently, each animal was placed in the microPET scanner under halothane gas anesthesia (5% induction and 1.5% for maintenance). A 15 min static acquisition was performed in the 3D mode. Data were collected in the list mode and reconstructed by a maximum a posteriori probability algorithm with a pixel size of 0.4 × 0.4 × 1.2 mm3.
PET data analysis
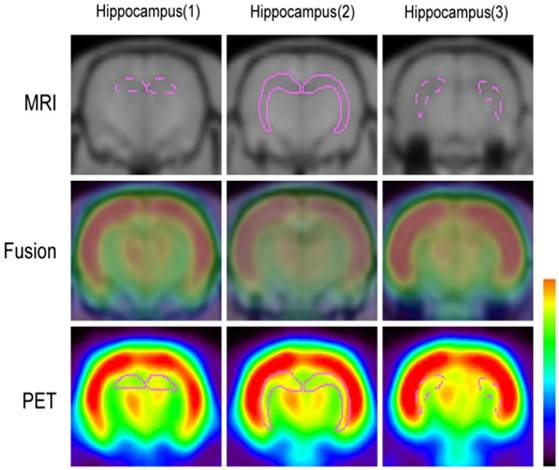
The data acquired from microPET images were displayed and analyzed by IDL (Version. 6.2, Research Systems, Colorado, USA) and ASIPro VM software (6.0.5.0, Concorde Microsystems Inc., Knoxville, USA). The PET and 3D FSPGR T1-weighted MRI images were co-registered using ASIPro VM software (Fig. 1). Glucose uptake was calculated by drawing regions of interest (ROIs) in the bilateral hippocampus for three slices and included the dorsal and ventral hippocampus. A semi-quantitative method (standardized uptake value, SUV) was used to calculate the changes of uptake rate in whole brain and hippocampus at each time point.
Co-registration of microPET and MRI images. The ROIs were selected according to the anatomical structures of the rat brain and included the dorsal and ventral hippocampus (purple).
Immunohistochemistry
Briefly, rats were anesthetized and perfused with 4% paraformaldehyde in 0.1 M PBS. The brains were removed and kept in 4% paraformaldehyde for 6 h, then immersed in 30% sucrose at 4°C for 3-4 days. After embedding in a mixture of 25% sucrose and O.C.T.™ (Sakura Finetek, USA), 15 µm sections were taken on a cryostat. Avidin-biotin staining was used in immunohistochemical staining and hematoxylin was used for counter-staining. The primary antibodies were rabbit anti-glial fibrillary acidic protein (GFAP) (1:200, Sigma) and mouse anti-neuron-specific nuclear protein (NeuN) (1:200, Chemicon). Normal goat serum and the absence of primary antibodies were used as negative controls. The same brain sections of CA1 were imaged using a magnification of 400x. The numbers of positive cells in an area of 0.02 mm2 were counted. The data were presented at mean and standard error. Cell counts were performed by a technician who is blind to the experimental design. All measurements were repeated three times and the mean value was used.
Statistical analysis
The data were presented as the mean ± standard deviation (SD). A multiple comparison using the Bonferroni correction was applied in order to compare image signal intensities and immunoreactive cells among the different phases in each group. The correlations were analyzed using the Pearson correlation. Statistical significance was set at P < 0.05.
Results
Temporospatial changes in glucose uptake
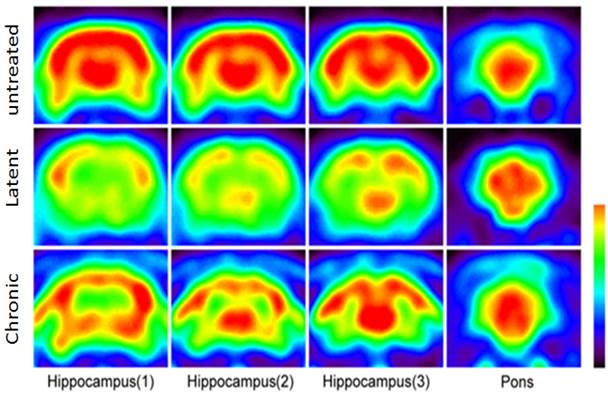
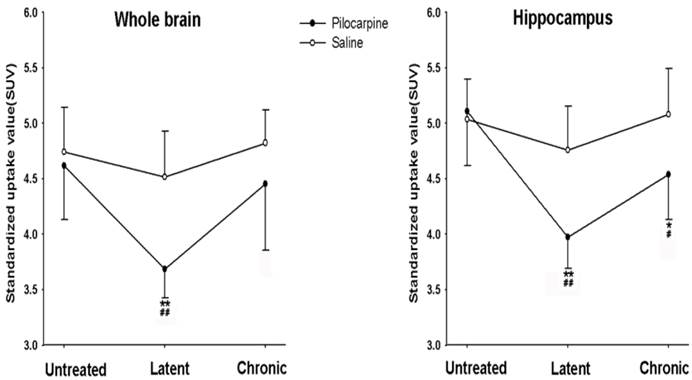
36 rats were initially included in this study. During the experiment, 12 of them were died and the other 24 rats were used in the analysis. SE had a significant effect on the 18F-FDG uptake in the whole brain and hippocampus when compared with untreated group (P < 0.01). Glucose uptake in the whole brain was significantly decreased in the latent phase (P < 0.01) and returned to the level obtained from untreated rats in the chronic phase (P > 0.05). 24 rats treated with saline were used as control. Saline did not affect the glucose uptake in the whole brain. Of note, the uptake value in latent phase after pilocarpine treatment was significantly lower than that in corresponding time point after saline treatment. The 18F-FDG-microPET images showed that the standardized uptake value (SUV) of 18F-FDG in the hippocampus was significantly decreased by 22.3% in the latent phase compared to the baseline (before pilocarpine injection) (P < 0.01). Glucose uptake in the hippocampus partially recovered in the chronic phase but was still below the untreated time point (P < 0.05, Fig. 2 and 3). Saline treatment did not affect SUV of 18F-FDG in the hippocampus. Compared with corresponding time points in control, pilocarpine treatment significantly decreased the SUV.
MRI co-registered microPET images of the coronal views in the different phases of epileptogenesis. Four of the 17 coronal views slices of the rat brain are shown, including the dorsal to ventral hippocampus and the pons.
Glucose uptake of the whole brain and hippocampus in the different phases after pilocarpine or saline injection. *P < 0.05; **P < 0.01 vs. the untreated phase; #P < 0.05; ##P < 0.01 between the pilocarpine and saline group.
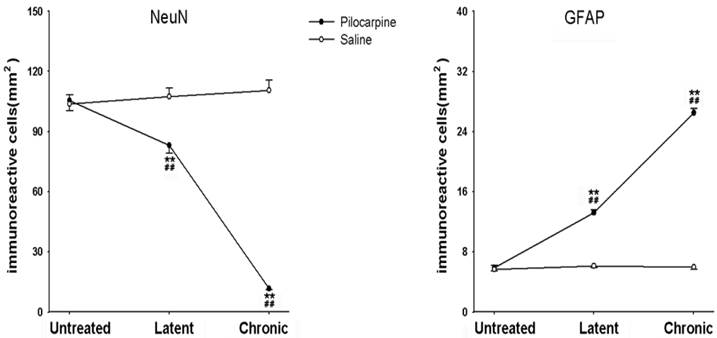
NeuN and GFAP immunohistochemistry in the hippocampus
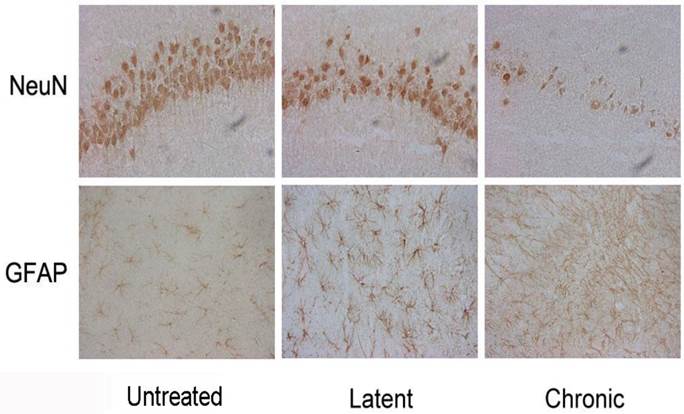
In the latent phase after pilocarpine injection, the numbers of NeuN positive cells in hippocampal CA1 area sections were significantly decreased compared to the untreated group (P < 0.01) and profound cell loss in the CA1 region (P < 0.01) were also noted. Conversely, GFAP-positive cells were significantly increased in the latent phase compared to the untreated group (P < 0.01) and significantly increased numbers of GFAP-positive cells were found in the hippocampus CA1 area in the chronic phase after pilocarpine injection (P < 0.01, Fig. 4 and 5). As the control, saline did not affect NeuN-positive cell numbers and GFAP-positive cell numbers.
Immunohistochemical staining for NeuN and GFAP in the different phases of epileptogenesis (×400).
NeuN- and GFAP-positive cells in the hippocampus in the different phases after pilocarpine or saline injection. *P < 0.05; **P < 0.01 vs. the untreated phase; #P < 0.05; ##P < 0.01 between the pilocarpine and saline group.
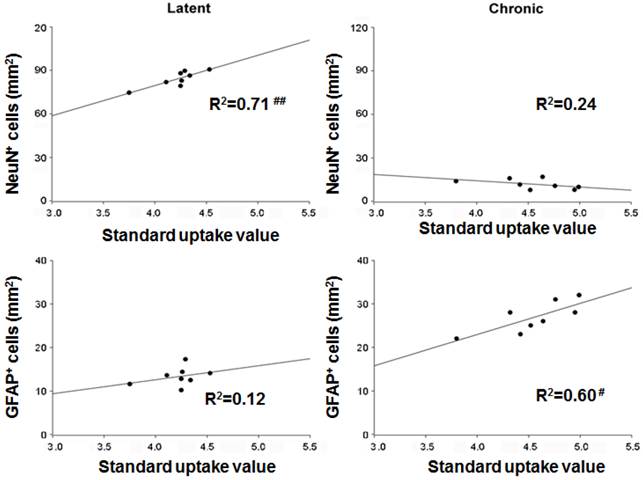
Correlation of hippocampal hypometabolism with NeuN and GFAP immunoreactivity
Using the Pearson correlation, the hippocampal glucose uptake correlated well with NeuN immunoreactivity in the latent phase (Pearson's product-moment coefficient, r = 0.84, P < 0.05) but not with GFAP immunoreactivity (P > 0.05). However, in the chronic phase there was a significant positive correlation between hippocampal glucose uptake and GFAP immunoreactivity (r = 0.78, P < 0.05) and no significant correlation was observed between hippocampal glucose uptake and NeuN immunoreactivity at this time point (P > 0.05, Fig. 6).
Hippocampal immunohistochemical staining and the correlation to glucose uptake in the different phases of epileptogenesis. #P < 0.05.
Discussion
Our study has demonstrated that hippocampal hypometabolism, visualized by 18F-FDG-microPET, is present in the early stages following SE, whereas there is partial glucose uptake recovery in the chronic phase. NSE and GFAP correlation analysis revealed that severe glucose hypometabolism correlated with neuronal cell loss in the latent phase and astrogliosis in the chronic phase.
In this study, we found glucose uptake in hippocampus was significantly decreased in the latent phase and there was a concurrent decrease in neuronal cell numbers. Previous studies have demonstrated widespread hypometabolism in the brain during the latent phase following SE and have additionally suggested that metabolic decreases that occur in damaged areas are due to neuronal cell loss [13-15]. Our findings are consistent with those studies. Hippocampal glucose uptake correlated significantly with NeuN immunoreactivity in the latent phase, suggesting the existence of a common mechanism underlying hypometabolism in these areas during this phase. Glucose uptake in the hippocampus was partly restored during the chronic phase; however, hippocampal cell loss continued to progress. Although the cell death already occurs in the first week after seizure in the latent phase [16], there was still cell degeneration likely caused by apoptosis and subsequent injury.
No correlation was observed between hippocampal glucose uptake and NeuN immunoreactivity during the chronic phase. This finding is in accordance with previous animal and clinical studies which also failed to demonstrate a significant correlation between hippocampal atrophy or neuronal density and glucose uptake in patients with TLE [17, 18]. Furthermore, patients without atrophy visible by MRI or significant surgically identifiable histologic cell loss often display marked temporal lobe hypometabolism [19]. These results suggest that hypometabolism is not entirely a consequence of neuronal cell loss. Instead, hypometabolism may promote the ongoing of epileptic network during epileptogenesis.
Widespread microglial activation and persistent astrogliosis in areas with neuronal death was found in pilocarpine-induced SE rats. These glial cell reactions develop during the first week after SE and persist into the chronic phase [20-22]. We observed that GFAP-positive cells are significantly increased in the latent phase and their numbers increase further in the chronic phase after pilocarpine injection. Our findings support the hypothesis that glial cells are activated after SE—a phenomenon that plays a crucial role in seizure generation or seizure spread in the chronic phase [23]. Astrocytes may also contribute to the metabolic signal. Previous studies have suggested that activated microglia may provide some component of the metabolic signature during acute seizures and in the peri-infarct zone of the rat model of ischemia using 18F-FDG-microPET [24, 25]. Recently, Lee et al. also found glial activation and synaptogenesis may reflect increased glucose uptake using 18F-FDG-microPET combined proton magnetic resonance spectroscopy (1 H MRS) in the rat lithium-pilocarpine model of epilepsy [26].Therefore, given the fact that the partial recovery of hippocampal glucose uptake was observed in the latent phase and the significant correlations between the magnitude of the hypometabolism and the changes in the levels of GFAP-positive cells, we postulate that the partial recovery of glucose uptake in the hippocampus may be attributable (in part) to gliosis without recovery of neuronal cell numbers during the epileptogenic process.
Nevertheless, there is limitation for our study. In our study, a standardized uptake value (SUV) with a semi-quantitative method was used to access the metabolic rate change of the whole brain and hippocampus. However, for the cell count, only CA1 region was chosen. That might lead to the discrepancy between the minor reduction of glucose consumption and vast neuronal death. In addition, high glycolytic rate in astrocytes possibly contributes to the discrepancy. This point deserves future study
Conclusion
Our study has outlined the specific temporospatial changes in glucose uptake that occur in the hippocampus during the course of epileptogenesis in the rat lithium-pilocarpine model of epilepsy. In addition, we have determined a possible underlying pathophysiological basis of these changes. Our results have revealed that severe glucose hypometabolism in hippocampus during the latent phase correlates with neuronal cell loss and the partial recovery of hippocampal glucose uptake in the chronic phase may be due to astrogliosis.
Acknowledgements
This study was supported by a grant from the National Science Foundation of China (NSFC) (No. 81271624, 81000556, 81300991).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Boling WW, Lancaster M, Kraszpulski M, Palade A, Marano G, Puce A. Fluorodeoxyglucose-positron emission tomographic imaging for the diagnosis of mesial temporal lobe epilepsy. Neurosurgery. 2008;63:1130-8 discussion 8
2. O'Brien TJ, Miles K, Ware R, Cook MJ, Binns DS, Hicks RJ. The cost-effective use of 18F-FDG PET in the presurgical evaluation of medically refractory focal epilepsy. J Nucl Med. 2008;49:931-7
3. Vinton AB, Carne R, Hicks RJ, Desmond PM, Kilpatrick C, Kaye AH. et al. The extent of resection of FDG-PET hypometabolism relates to outcome of temporal lobectomy. Brain. 2007;130:548-60
4. Chen S, Buckmaster PS. Stereological analysis of forebrain regions in kainate-treated epileptic rats. Brain Res. 2005;1057:141-52
5. Sharma AK, Jordan WH, Reams RY, Hall DG, Snyder PW. Temporal profile of clinical signs and histopathologic changes in an F-344 rat model of kainic acid-induced mesial temporal lobe epilepsy. Toxicol Pathol. 2008;36:932-43
6. Joo EY, Hong SB, Han HJ, Tae WS, Kim JH, Han SJ. et al. Postoperative alteration of cerebral glucose metabolism in mesial temporal lobe epilepsy. Brain. 2005;128:1802-10
7. Kuhl DE, Engel J Jr, Phelps ME, Kowell AP. Epileptic patterns of local cerebral metabolism and perfusion in man: investigation by emission computed tomography of 18F-fluorodeoxyglucose and 13N-ammonia. Trans Am Neurol Assoc. 1978;103:52-3
8. Guo Y, Gao F, Wang S, Ding Y, Zhang H, Wang J. et al. In vivo mapping of temporospatial changes in glucose utilization in rat brain during epileptogenesis: an 18F-fluorodeoxyglucose-small animal positron emission tomography study. Neuroscience. 2009;162:972-9
9. Tooyama I, Bellier J-P, Park M, Minnasch P, Uemura S, Hisano T. et al. Morphologic Study of Neuronal Death, Glial Activation, and Progenitor Cell Division in the Hippocampus of Rat Models of Epilepsy. Epilepsia. 2002;43:39-43
10. Gao F, Guo Y, Zhang H, Wang S, Wang J, Wu JM. et al. Anterior thalamic nucleus stimulation modulates regional cerebral metabolism: an FDG-MicroPET study in rats. Neurobiol Dis. 2009;34:477-83
11. Gao F, Wang S, Guo Y, Wang J, Lou M, Wu J. et al. Protective effects of repetitive transcranial magnetic stimulation in a rat model of transient cerebral ischaemia: a microPET study. Eur J Nucl Med Mol Imaging. 2010;37:954-61
12. Matsumura A, Mizokawa S, Tanaka M, Wada Y, Nozaki S, Nakamura F. et al. Assessment of microPET performance in analyzing the rat brain under different types of anesthesia: comparison between quantitative data obtained with microPET and ex vivo autoradiography. Neuroimage. 2003;20:2040-50
13. Andre V, Dube C, Francois J, Leroy C, Rigoulot MA, Roch C. et al. Pathogenesis and pharmacology of epilepsy in the lithium-pilocarpine model. Epilepsia. 2007;48(Suppl 5):41-7
14. Dube C, Boyet S, Marescaux C, Nehlig A. Relationship between neuronal loss and interictal glucose metabolism during the chronic phase of the lithium-pilocarpine model of epilepsy in the immature and adult rat. Exp Neurol. 2001;167:227-41
15. Jupp B, Williams J, Binns D, Hicks RJ, Cardamone L, Jones N. et al. Hypometabolism precedes limbic atrophy and spontaneous recurrent seizures in a rat model of TLE. Epilepsia. 2012;53:1233-44
16. Bischoff V, Deogracias R, Poirier F, Barde YA. Seizure-induced neuronal death is suppressed in the absence of the endogenous lectin Galectin-1. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:15590-600
17. O'Brien TJ, Newton MR, Cook MJ, Berlangieri SU, Kilpatrick C, Morris K. et al. Hippocampal atrophy is not a major determinant of regional hypometabolism in temporal lobe epilepsy. Epilepsia. 1997;38:74-80
18. Theodore WH, Gaillard WD, De Carli C, Bhatia S, Hatta J. Hippocampal volume and glucose metabolism in temporal lobe epileptic foci. Epilepsia. 2001;42:130-2
19. Carne RP, O'Brien TJ, Kilpatrick CJ, MacGregor LR, Hicks RJ, Murphy MA. et al. MRI-negative PET-positive temporal lobe epilepsy: a distinct surgically remediable syndrome. Brain. 2004;127:2276-85
20. Represa A, Niquet J, Pollard H, Ben-Ari Y. Cell death, gliosis, and synaptic remodeling in the hippocampus of epileptic rats. J Neurobiol. 1995;26:413-25
21. Belluardo N, Mudo G, Jiang XH, Condorelli DF. Induction of astroglial gene expression by experimental seizures in the rat: spatio-temporal patterns of the early stages. Glia. 1996;16:174-86
22. Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH. et al. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182:21-34
23. Seifert G, Carmignoto G, Steinhauser C. Astrocyte dysfunction in epilepsy. Brain Res Rev. 2010;63:212-21
24. Mirrione MM, Konomos DK, Gravanis I, Dewey SL, Aguzzi A, Heppner FL. et al. Microglial ablation and lipopolysaccharide preconditioning affects pilocarpine-induced seizures in mice. Neurobiol Dis. 2010;39:85-97
25. Schroeter M, Dennin MA, Walberer M, Backes H, Neumaier B, Fink GR. et al. Neuroinflammation extends brain tissue at risk to vital peri-infarct tissue: a double tracer [11C]PK11195- and [18F]FDG-PET study. J Cereb Blood Flow Metab. 2009;29:1216-25
26. Lee EM, Park GY, Im KC, Kim ST, Woo CW, Chung JH. et al. Changes in glucose metabolism and metabolites during the epileptogenic process in the lithium-pilocarpine model of epilepsy. Epilepsia. 2012;53:860-9
Author contact
![]() Corresponding author: Feng Gao, 88 Jiefang Road, Hangzhou, Zhejiang, China, 310009 Tel: +86571-87784711 Email: gaofeng7607com.
Corresponding author: Feng Gao, 88 Jiefang Road, Hangzhou, Zhejiang, China, 310009 Tel: +86571-87784711 Email: gaofeng7607com.