Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(3):243-247. doi:10.7150/ijms.10953 This issue Cite
Research Paper
Genetic Interaction Analysis of TCF7L2 for Biochemical Recurrence after Radical Prostatectomy in Localized Prostate Cancer
Chien-Shu Chen1, Chao-Yuan Huang2, Shu-Pin Huang3,4, Victor C. Lin5,6, Chia-Cheng Yu7,8,9, Ta-Yuan Chang10, Bo-Ying Bao1,11,12 ![]()
1. Department of Pharmacy, China Medical University, Taichung, Taiwan
2. Department of Urology, National Taiwan University Hospital, College of Medicine, National Taiwan University, Taipei, Taiwan
3. Department of Urology, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
4. Department of Urology, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
5. Department of Urology, E-Da Hospital, Kaohsiung, Taiwan
6. School of Medicine for International Students, I-Shou University, Kaohsiung, Taiwan
7. Division of Urology, Department of Surgery, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan
8. Department of Urology, School of Medicine, National Yang-Ming University, Taipei, Taiwan
9. Department of Pharmacy, Tajen University, Pingtung, Taiwan
10. Department of Occupational Safety and Health, China Medical University, Taichung, Taiwan
11. Sex Hormone Research Center, China Medical University Hospital, Taichung, Taiwan
12. Department of Nursing, Asia University, Taichung, Taiwan
Received 2014-10-31; Accepted 2015-1-12; Published 2015-2-5
Abstract
Backgroud: Accumulated evidence has demonstrated a significant role of the Wnt pathway in human prostate cancer. We hypothesize that genetic variants in the Wnt pathway effector, Transcription factor 7-like 2 (TCF7L2), may influence clinical outcomes in prostate cancer.
Methods: We comprehensively selected 12 tagged single-nucleotide polymorphisms (SNPs) to capture majority of common variants across TCF7L2, and genotyped in 458 localized prostate cancer patients treated with radical prostatectomy (RP). Kaplan-Meier analysis, Cox proportional hazard model, and survival tree analyses were performed to identify significant SNPs that correlated with biochemical recurrence (BCR) after surgery.
Results: A higher-order SNP-SNP interaction profile consisting of TCF7L2 rs7094463, rs10749127, and rs11196224 was significantly associated with BCR (Ptrend = 0.001). After adjusting for possible confounders, the genetic profile remained significant (Ptrend = 0.007). None of the studied SNPs were individually associated with BCR.
Conclusions: Our results support a genetic interaction in the TCF7L2 SNPs as a predictor of disease recurrence after curative RP in localized prostate cancer patients.
Keywords: biochemical recurrence, prostate cancer, radical prostatectomy, single-nucleotide polymorphism, TCF7L2, Wnt pathway
Introduction
Transcription factor 7-like 2 (TCF7L2), also known as TCF4, is an important effector of the canonical Wnt signaling pathway. Activation of the Wnt pathway leads to an increase of β-catenin stabilization in the cytoplasm and subsequent accumulation in the nucleus, where β-catenin acts as coactivator for the T cell factor/lymphoid enhancer-binding factor (TCF/LEF) transcription factor family to stimulate transcription of numerous target genes involved in cellular proliferation, apoptosis, and invasion [1]. Dysregulation of the Wnt pathway is highly associated with cancer initiation and progression, including prostate cancer [2]. The growth of both normal cells in the prostate gland and prostate cancer cells relies on androgen/androgen receptor (AR) signals. It was recently shown that AR is also a target gene for the TCF7L2/β-catenin complex [3]. Considering the crosstalk between Wnt and AR signaling pathways, together with the growth regulatory role of TCF7L2, we evaluated the influence of the genetic variants in TCF7L2 on disease recurrence in localized prostate cancer patients receiving curative radical prostatectomy (RP).
Materials and Methods
Patient recruitment and data collection
This study included 458 Taiwanese patients who underwent RP as initial therapy for localized prostate cancer, as described previously [4-7]. Briefly, patients diagnosed with histologically confirmed prostate cancer were recruited from four medical centers in Taiwan: National Taiwan University Hospital, Kaohsiung Medical University Hospital, E-Da Hospital, and Kaohsiung Veterans General Hospital. Demographic, clinical, and follow-up data were obtained from the medical records. Biochemical recurrence (BCR) was defined as two consecutive prostate-specific antigen (PSA) values of at least 0.2 ng/mL [8, 9]. All participants provided written consent, and the local ethics committees approved the research protocol.
Single-nucleotide polymorphisms (SNP) selection and genotyping
Genomic DNA was extracted from peripheral blood with the QIAamp DNA Blood Maxi Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol, and stored until the time of study. We utilized a tagging SNP approach to investigate all the genetic variability in the TCF7L2. Tagging SNPs were selected using the Tagger algorithm available through Haploview, using pairwise SNP selection with r2 ≥0.8 and minor-allele frequencies (MAF) ≥0.2 from the HapMap population data for Han Chinese in Beijing, China (CHB) [10, 11]. We identified 15 tagging SNPs for TCF7L2, but three SNPs that failed at Sequenom assay design were excluded. Genotyping was carried out at the National Center for Genome Medicine, Taiwan, using the Sequenom iPLEX matrix-assisted laser desorption/ionization time-of-flight mass-spectrometry technology. All 12 SNPs were in Hardy-Weinberg equilibrium (P > 0.05) and had average genotyping call rate ≥0.93. For quality control, we randomly selected 10 samples for duplicates, and the concordance rate was >0.99 for all SNPs assayed.
Statistical analysis
Patient clinicopathologic characteristics were summarized as either the numbers and percentages of patients, or the median and interquartile range of values. The association between patient characteristics with BCR was assessed by the log-rank test. Individual SNPs were initially assessed using the log-rank test for the three genetic models of inheritance: dominant (common homozygotes versus variant allele carrying genotypes), recessive (common allele carrying genotypes versus variant homozygotes), and additive (P for trend). Higher order SNP-SNP interactions were evaluated using survival tree analysis by STREE software (http://c2s2.yale.edu/software/stree/), which uses recursive partitioning to identify subgroups of patients with similar risk of disease recurrence [12]. Kaplan-Meier analysis with log-rank test was then used to estimate the survivals between each of the terminal subgroups and categorized into low-, medium-, and high-risk groups. Multivariate Cox proportional hazards regression analyses were used to assess the effect of genetic interaction profile in TCF7L2 on BCR, with or without adjusting for known prognostic factors, including age, PSA at diagnosis, pathologic Gleason score, stage, surgical margin, and lymph node metastasis, as previously described [5]. The Statistical Package for the Social Sciences software, version 22.0.0 (IBM, Armonk, NY, USA), was used for other statistical analyses. A two-sided P value of <0.05 was considered statistically significant.
Bioinformatics analysis
HaploReg v2 [13] and the Encyclopedia of DNA Elements (ENCODE) [14] data were used to identify the regulatory potential of the region adjoining the SNPs.
Results
We identified 184 (40.2%) patients experienced BCR in localized prostate cancer receiving RP during a median follow-up time of 54 months (Table 1). PSA at diagnosis, pathologic Gleason score, stage, surgical margin, and lymph node metastasis, were all significantly associated with BCR (P < 0.001).
Clinical characteristics of the study cohort
| Characteristic | P* | |
|---|---|---|
| Patients, n | 458 | |
| Age at diagnosis | 0.517 | |
| Median, y (IQR) | 66 (61-70) | |
| ≤66 | 241 (52.6) | |
| >66 | 217 (47.4) | |
| PSA at diagnosis | <0.001 | |
| Median, ng/mL (IQR) | 11.1 (7.1-17.5) | |
| ≤10 | 197 (44.9) | |
| >10 | 242 (55.1) | |
| Pathologic Gleason score, n (%) | <0.001 | |
| ≤7 | 392 (86.5) | |
| >7 | 61 (13.5) | |
| Pathologic stage, n (%) | <0.001 | |
| T1/T2 | 303 (67.2) | |
| T3/T4/N1 | 148 (32.8) | |
| Surgical margin | <0.001 | |
| Negative | 241 (72.6) | |
| Positive | 91 (27.4) | |
| Lymph node metastasis | <0.001 | |
| Negative | 433 (95.6) | |
| Positive | 20 (4.4) | |
| Recurrence | 184 (40.2) | |
| Median follow-up time†, mo (95% CI) | 54 (50-58) |
Abbreviations: IQR, interquartile range; PSA, prostate-specific antigen; CI, confidence interval.
*P value was calculated by the log-rank test for disease recurrence.
†Median follow-up time and 95% CIs were estimated with the reverse Kaplan-Meier method.
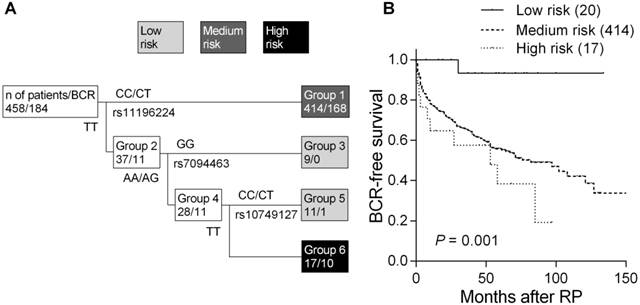
Of the 12 tagged SNPs we analyzed to capture majority of the variants in TCF7L2, none of the SNPs showed noteworthy associations with BCR under additive, dominant, and recessive models using the log-rank tests (all P > 0.05, Supplementary Material: Table S1). Therefore, higher order SNP-SNP interactions in modulating the risk of disease recurrence were further explored by survival tree analysis. We identified three tagged SNPs, rs11196224, rs7094463, and rs10749127, potentially having interactions, and the resulting tree structure was comprised of four terminal groups with low-, medium-, and high-risk of BCR according to the log-rank tests (Figure 1A). The median BCR-free survival of patients at low risk has not been reached during the follow-up. In comparison, median survival time was 82 months in the medium-risk group and the hazard ratio (HR) was 10.5 [95% confidence interval (CI) 1.47-75.0, P = 0.019, Table 2 and Figure 1B]. The median survival time was only 53 months for high-risk patients and the HR was 16.1 (95% CI 2.06-125, P = 0.008; P for trend = 0.001).
Higher order SNP-SNP interactions among TCF7L2 polymorphisms for BCR in localized prostate cancer patients. (A) Survival tree analysis identifies the interactions among the three polymorphisms. (B) Kaplan-Meier curves of BCR-free survival based on the survival tree analysis. Numbers in parentheses indicate the number of patients.
Cox proportional hazards analysis of TCF7L2 genetic interaction profiles with BCR in localized prostate cancer patients treated with RP
| Groups | n (%) | n of events | Median, mo | P | Univariate analysis | Multivariate analysis* | ||
|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |||||
| Low risk | 20 (4.4) | 1 | NR | 0.001 | 1.00 | 1.00 | ||
| Medium risk | 414 (91.8) | 168 | 82 | 10.5 (1.47-75.0) | 0.019 | 6.29 (0.87-45.4) | 0.068 | |
| High risk | 17 (3.8) | 10 | 53 | 16.1 (2.06-125) | 0.008 | 10.5 (1.34-83.0) | 0.025 | |
| Trend | 2.18 (1.38-3.47) | 0.001 | 2.08 (1.23-3.53) | 0.007 | ||||
Abbreviations: BCR, biochemical recurrence; RP, radical prostatectomy; NR, not reached; HR, hazard ratio; CI, confidence interval.
*HRs were adjusted for age, PSA, Gleason score, stage, surgical margin, and lymph node metastasis.
P < 0.05 are in boldface.
In multivariate analysis, adjusting for age at diagnosis, PSA, pathologic Gleason score, stage, surgical margin, and lymph node metastasis, the genetic interaction profile remained significant. In comparison to the low-risk group, the medium-risk group presented a 6.29-fold increased risk of disease progression (95% CI 0.87-45.4, P = 0.068, Table 2), and the high-risk group had a 10.5-fold increased risk (95% CI 1.34-83.0, P = 0.025; P for trend = 0.007). These data indicated that the genetic interaction profile in TCF7L2 provided additional predictive information beyond the conventional risk factors to influence prostate cancer outcomes.
Functional annotations from the ENCODE data for all correlated variants within the linkage disequilibrium (LD) blocks (r2 >0.8) containing the three interacting SNPs, rs7094463, rs10749127, and rs11196224, are shown in Figure 2. The rs7094463 and seven additional linked SNPs are situated at a locus with histone modification patterns characteristic of promoter in several cell types. In addition, the regulatory motif of forkhead box (Fox) transcription factors was predicted to be altered by rs7094463 (Figure 2 and Supplementary Material: Table S2). The tagged SNP rs10749127 and several rs11196224-linked SNPs are situated at a locus with histone modification patterns characteristic of enhancers, and possibly alter multiple regulatory motifs (Figure 2 and Supplementary Material: Table S3 and S4). This suggests that these variants are theoretically functional and might explain the association of TCF7L2 with disease progression.
Expanded view of the ENCODE data for the LD blocks containing the three interacting SNPs. ENCODE data showed evidence of promoter/enhancer elements coinciding with the variants linked with the three interacting SNPs, rs7094463, rs10749127, and rs11196224. The H3K4Me1, H3K4Me3, and H3K27Ac tracks show the genome-wide levels of enrichment of the mono-methylation of lysine 4, tri-methylation of lysine 4, and acetylation of lysine 27 of the H3 histone protein, as determined by the ChIP-seq assays. These levels are thought to be associated with enhancer and promoter regions. Chromatin State Segmentation track displays chromatin state segmentations by integrating ChIP-seq data using a Hidden Markov Model for eight different cell types. The chromatin state regions predicted for promoters and enhancers are highlighted. DNase clusters track shows DNase hypersensitivity areas. Tnx Factor track shows regions of transcription factor binding of DNA, as assayed by ChIP-seq experiments.

Discussion
TCF7L2 is the effector of the Wnt signaling pathway, and it combines with β-catenin to form an active nuclear complex that induces the expression of target genes involved in cellular proliferation, apoptosis, and invasion. Many studies have identified the association between TCF7L2 SNPs, rs7903146 or rs12255372 (in LD with each other), and the risk of several types of cancer [15-18], including prostate cancer [19]. However, these SNPs have a MAF of <0.05 in Asian populations, compared to >0.25 in other ethnic groups, thus limiting power to detect an association in Asian patients. In this study, we comprehensively selected 12 tagged SNPs to capture common genetic variability (MAF ≥0.2) in the TCF7L2, and determined their prognostic values. We showed that a genetic interaction profile consisting of the TCF7L2 rs7094463, rs10749127, and rs11196224 correlates with disease recurrence in prostate cancer patients treated with RP. The genetic interaction analysis takes the complexity of interacting SNPs into account, and this approach provides a novel way to use genetic variants in TCF7L2 to predict prostate cancer outcomes.
ENCODE data indicated that rs7094463 is located at a locus with histone modification patterns characteristic of promoter and a RNA polymerase II binding region (Figure 2 and Supplementary Material: Table S2). rs10749127 and rs11196224 coincide with regions of open chromatin, which probably correspond to the enhancers of TCF7L2 (Figure 2 and Supplementary Material: Table S3 and S4). In addition, multiple regulatory motifs were predicted to be altered by these SNPs. Therefore, it is plausible that these SNPs might influence TCF7L2 expression by altering the transcription factor binding sites. Further mechanistic studies are necessary to determine whether these significant SNPs have functional activity in the Wnt pathway or in the clinical outcome of prostate cancer patients.
In conclusion, this is the first study to explore the interaction between TCF7L2 SNPs in relation to the clinical outcomes for prostate cancer. The genetic interaction analysis relies on the data mining to identify the best model for the data, potentially leading to that the optimal results only showed in the initial test cohort. However, testing for the interaction among SNPs is probably more rational than testing for each individual SNP since the response to treatment is a complex phenomenon. If validated in independent studies, our results might be applicable to future modeling of clinical outcomes for prediction of disease recurrence in localized prostate cancer patients.
Supplementary Material
Table S1 -Table S4.
Abbreviations
TCF7L2, transcription factor 7-like 2; BCR, biochemical recurrence; RP, radical prostatectomy; SNP, single-nucleotide polymorphism; HR, hazard ratio; CI, confidence interval; PSA, prostate-specific antigen.
Acknowledgements
This work was supported by the Ministry of Science and Technology of Taiwan (grant number: 100-2314-B-039-009-MY3, 102-2628-B-039-005-MY3, and 103-2314-B-037-060), the China Medical University (grant number: CMU103-BC-5), and the Kaohsiung Medical University Hospital (grant number: KMUH-101-1R44 and KMUH103-3R43). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Chao-Shih Chen for data analysis and the National Center for Genome Medicine, Ministry of Science and Technology of Taiwan, for technical support. The results published here are based in part on data generated by the ENCODE and HaploReg projects.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843-50
2. Iozzo RV, Eichstetter I, Danielson KG. Aberrant expression of the growth factor Wnt-5A in human malignancy. Cancer Res. 1995;55:3495-9
3. Yang X, Chen MW, Terry S. et al. Complex regulation of human androgen receptor expression by Wnt signaling in prostate cancer cells. Oncogene. 2006;25:3436-44
4. Bao BY, Pao JB, Lin VC. et al. Individual and cumulative association of prostate cancer susceptibility variants with clinicopathologic characteristics of the disease. Clin Chim Acta. 2010;411:1232-7
5. Huang SP, Huang LC, Ting WC. et al. Prognostic significance of prostate cancer susceptibility variants on prostate-specific antigen recurrence after radical prostatectomy. Cancer Epidemiol Biomarkers Prev. 2009;18:3068-74
6. Huang SP, Lan YH, Lu TL. et al. Clinical significance of runt-related transcription factor 1 polymorphism in prostate cancer. BJU Int. 2011;107:486-92
7. Yu CC, Lin VC, Huang CY. et al. Prognostic significance of cyclin D1 polymorphisms on prostate-specific antigen recurrence after radical prostatectomy. Ann Surg Oncol. 2013;20(Suppl 3):S492-9
8. Freedland SJ, Sutter ME, Dorey F. et al. Defining the ideal cutpoint for determining PSA recurrence after radical prostatectomy. Prostate-specific antigen. Urology. 2003;61:365-9
9. Huang SP, Levesque E, Guillemette C. et al. Genetic variants in microRNAs and microRNA target sites predict biochemical recurrence after radical prostatectomy in localized prostate cancer. Int J Cancer. 2014;135:2661-7
10. de Bakker PI, Yelensky R, Pe'er I. et al. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217-23
11. International HapMap C, Frazer KA, Ballinger DG. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851-61
12. Zhang HP, Singer B. Recursive partitioning and applications. New York: Springer. 2010
13. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930-4
14. Rosenbloom KR, Sloan CA, Malladi VS. et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56-63
15. Burwinkel B, Shanmugam KS, Hemminki K. et al. Transcription factor 7-like 2 (TCF7L2) variant is associated with familial breast cancer risk: a case-control study. BMC Cancer. 2006;6:268
16. Folsom AR, Pankow JS, Peacock JM. et al. Variation in TCF7L2 and increased risk of colon cancer: the Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care. 2008;31:905-9
17. Goode EL, Szabo C, Prokunina-Olsson L. et al. No association between a candidate TCF7L2 variant and risk of breast or ovarian cancer. BMC Cancer. 2009;9:312
18. Hazra A, Fuchs CS, Chan AT. et al. Association of the TCF7L2 polymorphism with colorectal cancer and adenoma risk. Cancer Causes Control. 2008;19:975-80
19. Agalliu I, Suuriniemi M, Prokunina-Olsson L. et al. Evaluation of a variant in the transcription factor 7-like 2 (TCF7L2) gene and prostate cancer risk in a population-based study. Prostate. 2008;68:740-7
Author contact
![]() Corresponding author: Bo-Ying Bao, Department of Pharmacy, China Medical University, 91 Hsueh-Shih Road, Taichung 40402, Taiwan. Tel: +886-4-22053366 ext. 5126; Fax: +886-4-22031075; E-mail: baocmu.edu.tw
Corresponding author: Bo-Ying Bao, Department of Pharmacy, China Medical University, 91 Hsueh-Shih Road, Taichung 40402, Taiwan. Tel: +886-4-22053366 ext. 5126; Fax: +886-4-22031075; E-mail: baocmu.edu.tw