Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
A clinical overview of IDH1 in...
Physiological function of IDH1
Pathogenesis of IDH1 in...
Therapies targeting the IDH1...
Conclusions
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(3):201-213. doi:10.7150/ijms.11047 This issue Cite
Review
New Developments in the Pathogenesis and Therapeutic Targeting of the IDH1 Mutation in Glioma
Lilia Dimitrov1,2*, Christopher S. Hong2*, Chunzhang Yang2, Zhengping Zhuang2 ![]() , John D. Heiss2
, John D. Heiss2 ![]()
1. Barts and the London School of Medicine and Dentistry, Greater London, E1 2AD, United Kingdom
2. Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA
* Equal contribution
Received 2014-11-12; Accepted 2014-12-30; Published 2015-1-20
Abstract
In the last five years, IDH1 mutations in human malignancies have significantly shaped the diagnosis and management of cancer patients. Ongoing intense research efforts continue to alter our understanding of the role of the IDH1 mutation in tumor formation. Currently, evidence suggests the IDH1 mutation to be an early event in tumorigenesis with multiple downstream oncogenic consequences including maintenance of a hypermethylator phenotype, alterations in HIF signalling, and disruption of collagen maturation contributing to a cancer-promoting extracellular matrix. The most recent reports elucidating these mechanisms is described in this review with an emphasis on the pathogenesis of the IDH1 mutation in glioma. Conflicting findings from various studies are discussed, in order to highlight areas warranting further research. Finally, the latest progress in developing novel therapies against the IDH1 mutation is presented, including recent findings from ongoing phase 1 clinical trials and the exciting prospect of vaccine immunotherapy targeting the IDH1 mutant protein.
Keywords: IDH1 protein, glioma, DNA methylation, HIF1A protein, molecular targeted therapy, review
Introduction
Glioma is a broad term that includes primary malignant brain tumors of many types. Great effort has been expended to determine the genetic basis of these tumors, with the expectation that this knowledge will pave the way for the development of highly targeted therapies that will improve their generally poor prognosis.
Glioma has three main histological subtypes. Astrocytoma is the most common, accounting for 70% of all cases, while oligodendroglioma comprises 9%, and ependymoma 6% [1]. Tumors derived from mixed cell types make up most of the remaining cases. Glioblastoma (GBM) is the most malignant and most common type of astrocytoma, representing 55% of all cases of glioma. GBM treatment has traditionally involved surgery and radiation, with chemotherapy being of little additional value [2]. A recent randomized clinical trial demonstrated that the inclusion of temozolomide to surgery and radiotherapy resulted in a median survival of 15 months, 2.5 months more than surgery and radiation alone, and this regimen has become the current standard for GBM [3]. Life expectancy remains short, spurring additional research and development of more effective therapeutic strategies for GBM.
In 2008, The Cancer Genome Atlas (TCGA) conducted a genome-wide profile study, which identified, for the first time, mutations in the gene of isocitrate dehydrogenase 1 (IDH1) in GBM tumor samples [4]. The novel discovery in GBM of a mutation in a gene expressing an enzyme involved in cellular metabolism mirrored findings in non-central nervous system (CNS) tumors of mutation of genes expressing the metabolic enzymes succinate dehydrogenase and fumarate hydratase [5]. Since then, IDH1 mutations have been linked to other histopathological forms of glioma and to non-CNS malignancies.
This review describes the current role of IDH1 mutations in human malignancies, including glioma. IDH1 mutation-specific relationships with oncogenic signalling pathways are detailed to identify pathogenic events underlying tumor formation. Additionally, this update includes recent and ongoing therapies targeting the IDH1 mutant protein.
A clinical overview of IDH1 in human malignancy
Glioma
GBMs are divided into primary and secondary types. Both are histologically identical, so clinical features are used to distinguish them. Primary GBM is by far the more common, accounting for 80% of cases. It presents as a GBM and predominates in older adults. Secondary GBMs evolve from lower-grade tumors (grade II diffuse astrocytoma or grade III anaplastic astrocytoma) and are typically seen in younger patients [6].
In the landmark TCGA study, the authors sequenced 20,661 protein-coding genes in 22 primary and secondary GBM tumor samples and used high-density oligonucleotide arrays to look for amplifications and deletions. They found that five of the samples (22%) had a heterozygous missense mutation in the IDH1 gene, a single base substitution of guanine for adenine, leading to arginine substituting for histidine at codon site 132 (R132H) in the mutant IDH1 protein. Strikingly, this mutation was present in 5 of the 6 secondary GBMs but none of the 16 primary GBMs. A follow-up targeted sequence analysis of an additional 127 tumors found the same IDH1 mutation in 13 of the samples with 4/5 (80%) of the secondary GBM tumors demonstrating the IDH1 mutation. Overall, the IDH1 mutation was found in 12% of the 149 tumors that were analysed. In a recent literature review, the IDH1 mutation was found in 5.6% of primary GBMs analysed across all studies (75/1345 tumors), and in 76% (94/123 tumors) of secondary GBMs, supporting the original findings of the TCGA study [7]. The IDH1 mutation is also prevalent in lower grade gliomas, occurring in over 70% of grade II tumors [8], and 62-80% of grade II-III oligodendrogliomas, grade II-III oligoastrocytomas, and grade III astrocytomas [7].
The TCGA study also importantly found that IDH1 mutations were more frequent in younger patients. The median age of patients with tumors harboring IDH1 mutations was 33.2 years, starkly contrasting the median age of 55.3 years in patients with wild type tumors. This study also demonstrated that in GBM patients, the IDH1 mutation conferred a survival advantage compared to IDH1 wild type GBM, with a median overall survival of 3.8 years in the former and 1.1 years in the latter. This finding has been replicated in other studies, with a survival of 2.6 years in IDH1 mutated tumors compared to just 1.2 years in wild type IDH1 tumors [9]. It is unclear whether IDH1 status alone is responsible for this survival advantage or whether other characteristics of secondary GBM improve its prognosis over primary GBM. Recently, Beiko et al (2014) demonstrated that IDH1 mutations were associated with higher rates of total surgical resection of enhancing regions in grade III and IV astrocytomas [10]. Furthermore, maximal resection of total tumor volume, including non-enhancing areas, led to improved overall survival in IDH1 mutated tumors but not in wild type counterparts. These results suggest that greater amenability to complete surgical resection may contribute to the improved prognosis of patients with IDH1 mutated gliomas. Complete surgical resection of total tumor volume (enhancing and non-enhancing areas) may be of greater significance to patient prognosis in IDH1 mutated tumors, compared to wild types. Further investigations are required to elucidate additional mechanisms behind the improved survival seen in patients with IDH1 mutated gliomas. This may be aided by a study comparing the survival of patients with secondary gliomas having the IDH1 mutation with the survival of patients with secondary IDH1 wild type gliomas.
IDH1 represents a gene that shows differential expression between primary and secondary GBMs. PTEN loss, EGFR amplification, and loss of heterozygosity (LOH) of chromosome 10 are associated with primary GBM while ATRX mutations, loss of p53, and LOH of chromosome 19 are common in secondary GBM [6, 11-14]. However, the IDH1 mutation predicts secondary GBM better than these other mutations predict their respective GBM subtypes.
Extensive genomic profiling has identified that around 90% of IDH1 mutations involve the R132H substitution [15, 16]. There may be some selection pressure for R132H, as this mutation is associated with the lowest levels of the compound 2-hydroxyglutarate (2-HG), which is lethal at high doses [17]. Of the remaining 10% of IDH1 mutations, 4.3-4.7% are due to arginine being replaced with cysteine (R132C), 1.9-2.1% with glycine (R132G), 1.6-1.7% with serine (R132S), 0.6-0.8% with leucine (R132L), and 0.3% with glutamine (R132Q) [16, 18]. Although no studies have compared patient outcomes among different IDH1 R132 mutations, R132S- and R132L-transfected human embryonic kidney cells produce significantly higher levels of 2-HG and exhibit markedly reduced cell viabilities compared to R132H-transfected cells, in vitro [16]. In addition, the specific type of IDH1 mutation appears to correspond to distinct histological types suggesting functional differences between mutations. For example, R132C mutations occur more frequently in astrocytoma than in oligodendroglioma [19]. The type of other genetic mutations co-occurring with the IDH1 mutation also influences the histological type of glioma. For example, astrocytomas tend to feature IDH1 and TP53 mutations, while IDH1 mutated oligodendrogliomas frequently have co-deletions of chromosomes 1p and 19q [20].
Different patterns of the IDH1 mutation between primary and secondary GBM, as well as between other grades of astrocytomas, and between other types of gliomas, is very useful diagnostically, helping to differentiate between histological subtypes which can often be subject to human error [21]. As well as providing patients with more accurate diagnosis and prognosis, more precise characterization of molecular features could open the door to a whole host of new individualised treatments.
Non-CNS malignancies
IDH1 mutations are also present in some tumors originating in cells outside of the CNS. In a sample of 224 patients with acute myeloid leukaemia (AML), 9% of tumors possessed the IDH1 mutation [22]. IDH mutations are more prevalent in AML if IDH2 mutations are also considered, with rates between 15-33% [23-25]. IDH1/2 mutations have also been found in 5% of patients with myelodysplastic syndrome (MDS), 8.8% with myeloproliferative neoplasms (MPN) and just under 10% of patients with secondary AML [26]. Unlike in GBM, IDH mutations have a negative impact on prognosis in MPN and MDS [27].
In a study, over one-half of central chondrosarcomas, central chondromas, and periosteal chondromas displayed IDH1/2 mutations [28]. This link between IDH mutations and connective tissue tumors was reported by the same group that identified IDH mutations to occur in patients with Ollier disease and Maffucci syndrome. These mainly pediatric disorders are characterized by the development of multiple tumor types and by somatic mosaicism of the IDH1 mutation. The majority of Ollier disease and Maffucci syndrome patients exhibit the R132C IDH1 mutation, in contrast to most secondary GBMs, which harbour the R132H mutation [29]. Interestingly, both Ollier disease and Maffucci syndrome are associated with the development of benign cartilaginous tumors, AML, and gliomas [30]. In addition, 10% of cholangiocarcinomas harbor IDH1 or 2 mutations; the prognostic significance of the mutation in this malignancy is unknown [31]. Although less well documented, other CNS tumors including ganglioglioma and primitive neuroectodermal tumor have also been linked with the IDH1 mutation [32, 33].
Physiological function of IDH1
In humans, IDH occurs as 3 isozymes: Idh1, Idh2 and Idh3 [34]. These isozymes are encoded by five genes: IDH1, IDH2, IDH3A, IDH3B and IDH3G. All are metabolic enzymes expressed by eukaryotic cells that act on the substrate isocitrate, converting it to alpha-ketoglutarate (a-KG) via oxidative decarboxylation. The reactions catalysed by IDH1 and IDH2 are reversible and use NADP+ as an electron acceptor leading to the production of NADPH [35]. IDH1 acts in the cell cytoplasm and peroxisomes whereas IDH2 and IDH3 are found in the mitochondrial matrix. The formation of non-mitochondrial NADPH by IDH1 is thought to be an important mechanism for limiting cellular oxidative damage. NADPH also acts as a reducing agent in lipid biosynthesis [36, 37]. The product of the IDH1 forward reaction, a-KG, is an intermediate in the tricarboxylic acid cycle (TCA) and is also involved in nitrogen transportation, oxidation reactions, and amino acid formation. In conditions of hypoxia, the reverse reaction is favored, in which IDH1 catalyzes the conversion of a-KG to isocitrate which can in turn be converted to acetyl-CoA for lipid metabolism [38, 39]. Additionally, IDH1 regulates glucose-stimulated insulin secretion [40].
Pathogenesis of IDH1 in malignancy
Introduction
Mutations to IDH1 appear to occur early on in glioma development, preceding loss of chromosomes 1p and 19q [40]. From a total of 321 biopsies taken over time from patients with grade II and III gliomas, there were no instances where TP53 mutations or 1p/19q co-deletions were found to develop prior to IDH1 mutation. This may be due to a strand asymmetrical mechanism, in which the IDH1 mutation is found on the template strand while TP53 mutations are on the coding strand and are thus only able to be transcribed after DNA replication [41].
Although the current understanding of IDH1 mutations in tumorigenesis remains incomplete, several important advances have been made that elucidate key molecular mechanisms. Unlike other metabolic enzymes associated with cancer such as fumarate hydratase and succinate dehydrogenase, the IDH1 mutation is a gain-of-function mutation, conferring neo-morphic activity upon IDH1 [4]. In a pivotal study profiling IDH1 wild type and mutant (R132H) glioma cells with liquid chromatography-mass spectrometry, Dang et al (2009) demonstrated that the mutant glioma cells express high levels of the metabolite 2-hydroxyglutarate (2-HG) [42]. Cellular levels of 2-HG in the wild type cells were usually below 0.1 mM, whereas levels in IDH1 mutated glioma cells reached 35 mM. The authors demonstrated that mutant IDH1 protein catalyzes the reduction of a-KG to the R-enantiomer of the metabolite, 2-HG (R-2-HG). Specifically, the mutation reduces the affinity of the IDH1 active site for isocitrate while concomitantly increasing it for NADPH and a-KG [43]. Reduced affinity for isocitrate occurs as a result of alterations to a binding site residue that forms hydrogen bonds between the alpha and beta carboxyl groups of isocitrate [43]. Consequently, the reverse reaction of IDH1 (a-KG to isocitrate) is favored but rather than carboxylate, the mutant enzyme reduces a-KG to form 2-HG (Fig. 1).
Of the three IDH isozymes, only IDH1 exists in the cytosol while IDH2 and IDH3 function within the mitochondria. Under normal conditions, cytosolic isocitrate is converted into a-KG by the wild type IDH1 enzyme with concurrent reduction of NADP+. Subsequently, a-KG can re-enter Kreb's cycle within the mitochondria or remain in the cytosol as an essential substrate for PHD. Among its many functions, in conditions of normoxia, PHD utilizes oxygen as a co-substrate and hydroxylates proline residues on HIF1-a, initiating proteasomal degradation via the VHL ubiquitin-ligase protein complex. Unlike its wild type counterpart, the mutant IDH1 protein exhibits neo-morphic activity and catalyzes conversion of a-KG into R-2-HG an “onco-metabolite” that promotes tumorigenesis through multiple pathways.
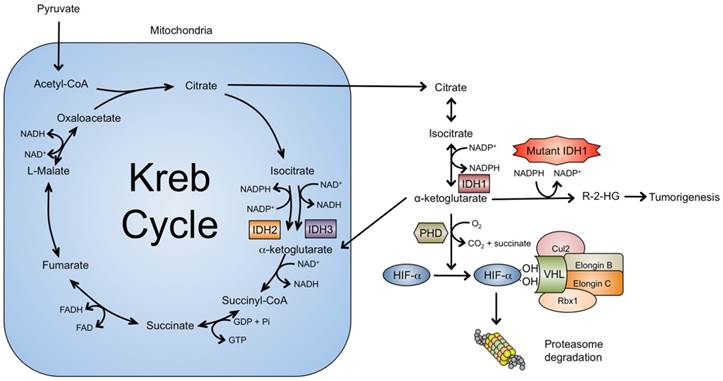
2-HG exists as two possible enantiomers, both of which occur physiologically as metabolic by-products [44]. In physiological conditions, the R-type is formed when gamma-hydroxybutyrate is converted to succinic semialdehyde while the S-type is formed during the conversion of oxaloacetate to L-malate in the TCA cycle [45, 46]. To date, only the R-enantiomer has been associated with IDH1 mutant proteins. Interestingly, R-2-HG formation catalyzed by mutant IDH1 requires heterozygosity of the IDH1 locus as homozygous IDH1 mutations show significantly reduced levels of R-2-HG [42, 47]. It has been suggested that mutant IDH1 may source a-KG produced by the wild type enzyme, contributing to high levels to R-2-HG [47]. This has been recapitulated by Brooks et al (2014) who demonstrated that the heterodimer of wild type and mutant IDH1 proteins had a Km approximately 11-fold lower than that of the mutant homodimer [48].
Several studies have shown that high levels of R-2-HG are able to mediate the changes seen in IDH1 mutants and as such, R-2-HG has been termed an “onco-metabolite” [49]. In an experiment using TF-1 leukemia cells, introduction of cell-permeable R-2-HG inhibited differentiation in response to erythropoietin (EPO) and induced growth factor resistance [27]. Both of these outcomes are important hallmarks in the formation of leukemia. This study demonstrated that continuously elevated levels of R-2-HG were needed to maintain tumor phenotype in IDH1 mutant cells as withdrawal of R-2-HG restored the normal differentiation response to EPO and growth factors. Further support for the role of 2-HG comes from the observation that patients with L-2-hydroxyglutaric aciduria, an inborn error of metabolism characterized by elevated levels of S-2-HG, have a higher risk of developing gliomas [50]. Interestingly however, patients with D-2-hydroxyglutaric aciduria, a similar metabolic disorder that is characterized by elevated R-2-HG, are not at increased risk for glioma or formation of other tumors [51]. The reason for this discrepancy is unclear and is an area requiring further investigation.
A number of potential mechanisms have been proposed to explain how R-2-HG produced by the mutant IDH1 protein promotes tumor formation. Epigenetic modification, via inhibition of a-KG-dependent dioxygenases leading to DNA and histone hypermethylation, has been at the forefront of research efforts [52]. Additional mechanisms implicated in tumor formation in IDH1 include inhibition of several groups of prolyl hydroxylases (PHD), leading to HIF1-a activation and alterations in collagen formation. Evidence for these findings is subsequently discussed in greater detail.
Targeting of hypoxia-inducible factors
Hypoxia-inducible factors (HIFs) are transcription factors that activate an array of genes important in the cellular response to hypoxia. Targeted downstream effects include angiogenesis, glucose metabolism and cell proliferation. HIF1 is a heterodimer made up of the HIF1-a and HIF1-b subunits, the former of which is active during hypoxic conditions but is unstable and degraded by von-Hippel Lindau protein (VHL) in the presence of oxygen. When oxygen levels are sufficient, the 2-KG-dependent PHD, Eg1N, hydroxylates proline residues on HIF1-a, creating a binding site for the VHL ubiquitin-ligase protein complex, which subsequently ubiquitinates HIF1-a for proteasomal degradation. In conditions of hypoxia, the HIF1-a PHD is inhibited as it requires oxygen as a co-substrate for enzymatic activity [53]. As such, HIF1-a degradation is circumvented and instead HIF1-a combines with the corresponding beta subunit, translocates to the nucleus, and activates target genes that facilitate cell survival in hypoxia and also may contribute to tumor formation (Fig. 2).
High levels of R-2-HG produced by the mutant IDH1 protein inhibit hydroxylation of HIF1-a by PHD. As such, HIF1-a persists, combines with the beta subunit, and translocates to the nucleus, where it induces transcription of hypoxia-related genes that may also promote oncogenic transformation and cell survival.
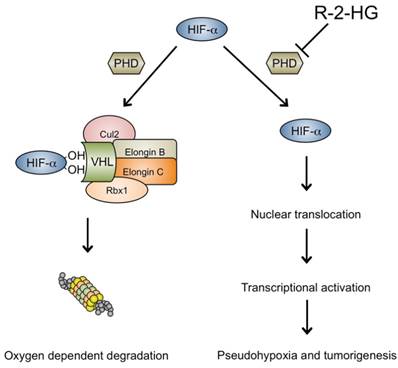
Considering that the Eg1N PHD is 2-KG-dependent, it was initially proposed that IDH1 mutations could cause tumor formation due to failure of HIF degradation secondary to impaired HIF1-a proline residue hydroxylation (Fig. 2) [54]. Increased levels of the transcription factor HIF1-a and its target genes have been found in the brain cells of IDH1 R132H knock-in mice [55]. More recently, it was shown that transfection of the IDH1 mutation into glioma cell lines upregulated HIF1-a and increased cell proliferation [56]. The authors suggested that this was mediated by transcriptional activity of HIF1-a dependent nuclear factor-κB (NF-κB) as mutant IDH1-mediated activation of NF-κB was abolished in a HIF1-a-dependent manner.
It is well established that HIF activation has an important role in tumor formation. However recent work suggests that the picture is more complex than this, with evidence that HIF1-a and HIF2-a have an antagonistic relationship [57]. In renal cell carcinoma, HIF1-a and HIF2-a have been shown to have tumor suppressive and promoting effects, respectively [58]. These observations have extended to IDH1 mutated glioma. In contrast to aforementioned studies demonstrating elevated levels of HIF1-a in IDH1 mutated glioma, other groups have found HIF1-a levels to be low. R-2-HG has been shown in astrocytes to act as a partial agonist for Eg1N, resulting in lower HIF levels but interestingly increased astrocyte proliferation [59]. The possibility that the IDH1 mutation drives cell proliferation via diminished HIF expression has been corroborated in several glioma studies. Williams et al (2011) looked at 120 human glioma samples and found that HIF1-a was only upregulated in a small subset of IDH1 mutated gliomas and was generally limited to necrotic areas [60]. Immunohistochemical analysis showed that in non-necrotic areas that were strongly reactive for the R132H IDH1 mutation, there was no evidence of HIF1-a overexpression. HIF upregulation in necrotic areas may explain the elevated levels of HIF1-a in the mouse model described by Sasaki et al, (2012) [55]. Mouse models of the IDH1 mutation have been associated with hemorrhage and high perinatal mortality and therefore it is difficult to exclude that the observed upregulation of HIF and corresponding target genes were not secondary to these events.
Undoubtedly, further work is needed to clarify the role of HIFs in IDH1 mutated glioma. Although traditionally considered as oncogenic, there is mounting evidence that HIFs have tumor suppressive properties in both CNS and hematologic malignancies [61]. As such, pharmacological inhibition of Eg1N activity has been proposed as a potential target for IDH1 mutant glioma and may be an important topic of future study [59].
Aberrant collagen maturation and stability
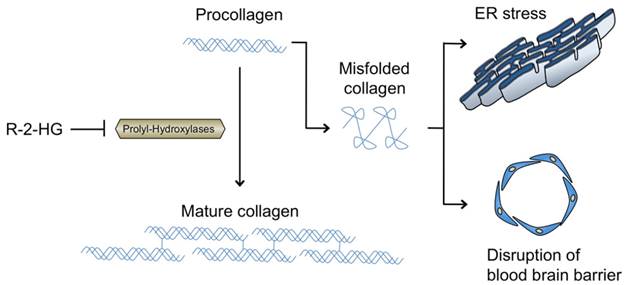
In addition to HIF regulation, PHDs are also involved in the post-translational modification of collagen, a process essential for collagen maturation and stability [62]. Three main a-KG-dependent PHD families are implicated in this activity: the leprecan prolyl-3-hydroxylases, the prolyl-4-hydroxylase alpha subunits, and the procollagen-lysine, 2-oxoglutarate 5-dioxygenase (PLOD) lysyl-5-hydroxylases. PHDs hydroxylate proline residues on type IV collagen, which is required for formation of the collagen triple helix whereas the lysyl-hydroxylates hydroxylate lysine residues that permit cross-linking between fibrils. Type IV collagen contributes to the integrity of blood brain barrier (BBB) and is specifically found in the basement membrane between astrocytes and endothelial cells. In the animal model of the IDH1 R132H mutation described earlier, mice were found to have higher levels of immature type IV collagen [55]. As a-KG-dependent post-translational changes to collagen occur in the endoplasmic reticulum (ER), it has been proposed that inhibition of a-KG by R-2-HG may cause accumulation of misfolded collagen in the ER, triggering an ER stress response that may contribute to the early lethality seen in IDH1 mutant embryos (Fig. 3). Additionally, impairment of perivascular type IV collagen may promote progression and breakdown of the physiological BBB in IDH1 mutated gliomas [55, 63, 64]. Given mutations in collagen synthesizing genes have been associated with IDH mutations in non-CNS tumors [65], future research may uncover similar findings in glioma as well as better define the role of IDH1 mutations in BBB disruption.
The hydroxylation of proline residues on pre-collagen fibrils by PHD is required for proper triple helix formation and maturation of type IV collagen. Disruption of PHD by R-2-HG produced by mutant IDH1 leads to accumulation of misfolded collagen, triggering a pro-apoptotic endoplasmic reticulum (ER) stress response. Additionally, as type IV collagen is found in the perivascular spaces of the brain, abnormal collagen build-up may contribute to breakdown of the blood brain barrier (BBB) in IDH1 mutated glioma.
A hypermethylator phenotype
DNA methylation, in particular CpG island hypermethylation, is a well-established hallmark of certain human cancers [66]. Methylation at these sites results in gene silencing, raising the possibility that tumor suppressor genes can be targets of this silencing and thus promote tumor formation. Recently, a quantitative analysis of the methylation status of five known tumor suppressor genes was performed in glioma cells and in glioma cell-free DNA from serum, which found that tumor methylation of PARP-1, SHP-1, DAPK-1 and TIMP-3 genes was positively correlated with tumor grade and negatively correlated with prognosis [67].
A subset of the 272 GBM tumors from TCGA and additional low-grade gliomas (LGG) analyzed for DNA methylation were found by Noushmehr et al (2010) to have overlapping methylated DNA loci, suggestive of a pattern of CpG island methylation [68]. The authors termed this the glioma-CpG island methylator phenotype (G-CIMP). They found that the G-CIMP phenotype was strongly associated with the IDH1 mutation and was more common in younger patients and associated with improved prognosis. Similar associations between global hypermethylation and IDH1/2 mutations have been observed in IDH1/2 mutated AML cells [69]. The G-CIMP phenotype has also recently been found to include tumor suppressive miRNAs with the finding that methylation of miR-148a is associated with IDH1 mutated glioma cells [70].
There is evidence to suggest that the association between hypermethylation and IDH1 mutations may be causal. Transfection of mutant IDH1 into immortalized primary human astrocytes resulted in the hypermethylator phenotype [71]. Similarly, introduction of ectopic mutant IDH1 into normal human astrocytes caused total genome hypermethylation as seen in IDH1 mutated LGG [72]. In the IDH1 mutation mouse model described by Sasaki et al (2012), mice with the mutant gene in the myeloid lineage alone had a similar hypermethylation pattern as seen in AML patients with IDH1/2 mutations and interestingly developed hematological malignancy-associated features of anemia, splenomegaly and extramedullary hematopoiesis [73]. More recently, Kernytsky and colleagues (2014) demonstrated that in vitro treatment with a small molecule inhibitor (AGI-6780) reversed histone and genomic DNA methylation patterns seen in an erythroleukemia model of IDH2 (R140Q) mutated TF-1 cells [74]. Importantly, the authors showed that exposure to AGI-6780 led to therapeutic demethylation of gene signatures that are known to be hypermethylated in hematologic malignancies. As such, further studies are required to corroborate whether genes hypermethylated by IDH1 and IDH2 mutations are indeed tumor suppressor genes. From a clinical standpoint, in order for G-CIMP to be useful prognostically, precise promoter loci must be defined.
IDH1 mutation-mediated silencing of TET2
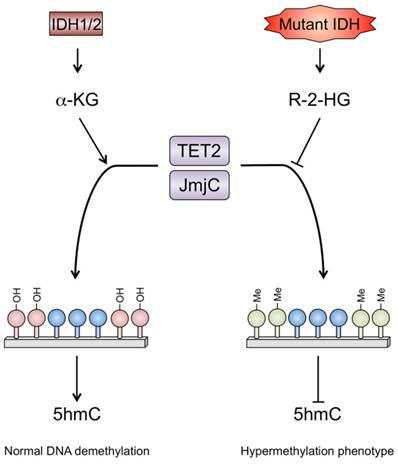
The leading mechanism attributed to the observed hypermethylation phenotype in IDH1 mutants involves silencing of the a-KG-dependent DNA modifying enzyme, Tet methylcytosine dioxygenase 2 (TET2). This myeloid tumor suppressor enzyme is one of three enzymes (TET1, TET2, TET3) dependent on a-KG to hydroxylate 5-methylcytosine (5mc) to 5-hydroxymethylcytosine (5hmc) during DNA demethylation [75]. It has been proposed that because R-2-HG is very similar structurally to a-KG, it may act as a direct competitive inhibitor of a-KG-dependent dioxygenases such as TET2 [59, 76, 77]. TET2 inhibition may encourage DNA hypermethylation through impaired DNA demethylation, leading to the hypermethylation phenotype (Fig. 4). TET2 has been a particular focus of research because it has been linked to hematological malignancies. Heterozygous loss-of-function TET2 mutations are seen in 10-25% of myeloid disorders such as AML, MDS, and chronic myelomonocytic leukemia (CMML) [78].
Under normal conditions, TET2 utilizes a-KG as a substrate to hydroxylate 5-methylcytosine (5mc) to 5-hydroxymethylcytosine (5hmc) during DNA demethylation. a-KG also binds to the JmjC domain of histone demethylases, which function to demethylate lysine residues on histone tails and subsequently regulate gene transcriptional activity. R-2-HG produced by the mutant IDH1 protein acts as a competitive inhibitor of TET2 and JmjC, promoting a hypermethylator phenotype that maintains an undifferentiated tumor state.
Genetic and epigenetic profiling of AML patients has revealed that TET2 mutated AML cells possess a hypermethylation signature that may contribute to impaired differentiation and elevation of stem cell markers [69]. However hypermethylation in response to loss of TET2 function has not been consistently found across studies. In fact several studies have reported the reverse pattern, with hypomethylation in TET2 mutated AML cells and hypermethylation in TET2 wild type cells [79, 80]. In another study, no difference in methylation was observed between wild type and mutant TET2 CMML cells [81]. As such, it is evident that although loss of TET2 is strongly linked to malignancy, the precise mechanism underlying this observation is undoubtedly still unclear [47]. Other factors likely contribute to whether loss of TET2 leads to a hypermethylator phenotype and tumor formation.
Experimental findings have been mixed in terms of the effect of IDH1 mutations on TET2 activity. Evidence of reduced levels of 5hmc in IDH1 mutated cells compared to wild types has been reproduced across several studies in glioma cells [71, 77] and in AML cells [69]. Transfection of TET2-expressing AML cells with the IDH1 mutation nearly halved 5hmc levels. Similarly, expression of the IDH1 and IDH2 mutations in cell lines derived from GBM led to reduced 5hmc levels whereas expression of IDH1 and TET1/2 wild types increased 5hmc levels, suggesting an inverse relationship between the IDH1 mutation and 5hmc levels. In the IDH1 mutant mouse model, mice expressing the IDH1 mutation in brain cells alone were found to have lower levels of 5hmc [55].
Perhaps, the most significant study demonstrating that the tumorigenic effects of the IDH1 mutation arise due to TET2 dysfunction was the discovery that IDH1/2 and TET2 mutations were mutually exclusive in 300 AML samples [69]. Furthermore, similar methylation signatures were found in IDH1 and TET2 mutants, involving over 60% of the genetic loci, suggesting overlapping effects between the two mutations. However, these results have not been replicated in glioma where one group demonstrated that none of 35 IDH1 wild type LGGs was found to have TET2 mutations [82]. Interestingly, however, the IDH1 wild types were associated with TET2 promoter methylation, which was not the case for any of the 38 IDH1 mutated LGGs. This finding suggested that IDH1 mutations and TET2 methylation could be mutually exclusive, with TET2 methylation providing an alternative mechanism for tumorigenesis in IDH1 wild type LGG. Exclusivity of IDH1 and TET2 mutations in leukemia has been suggested to result from a clonal disadvantage of IDH1 mutations for TET2 mutants [27]. Future studies may elucidate whether the same holds true in glioma.
On the other hand, numerous groups have reported findings against inhibition of TET2 by R-2-HG. Muller et al (2012) found that 61% of gliomas (wild type and IDH1 mutants) showed non-existent levels of 5hmc whereas high levels of 5hmc were found in 33% of IDH1 mutants [83]. Low 5hmc levels were associated with nuclear exclusion of TET1, perhaps by the promoter methylation mechanism observed by Kim et al (2011) [82]. Interestingly nuclear exclusion occurred more frequently in IDH1 wild types whereas IDH1 mutant gliomas were associated with nuclear accumulation of TET1. Although this counteracts a R-2-HG-mediated inhibition of TET, this study focused on TET1, which has been far less studied in human malignancy compared to TET2. Secondly, TET2 knockout and IDH1 gain-of-function mouse models have been shown to differ to a wide extent phenotypically, suggesting these mutations may contribute to tumor formation in a parallel rather than in a cooperative manner.
Histone hypermethylation and the Jumonji transcription factor family
The four histone proteins H2A, H2B, H3 and H4 have an important scaffolding role for DNA, packaging it into structural units called nucleosomes [84]. Histone tails are the sites at which numerous modification reactions occur, with histone tail methylation being a major focus of current cancer research. Histone methylation is important for modifying chromosome structure and can either activate or inhibit transcription of associated genes. For example, methylation of the histone residues H3K4, H3K36 and H3K79 activate euchromatin for transcription whereas the reverse is true for residues H3K9, H3K27, and H4K20 [85, 86]. Histone methylation is tightly controlled by a balance between histone methyltransferases and histone demethylases with the latter removing methyl groups from lysine residues on histone tails that are mono-, bi-, and tri-methylated. Alterations of this fine balance have significant effects on gene expression [87].
There is evidence that certain histone demethylases may act as tumor suppressors, with inhibition of specific histone demethylases implicated in clear cell renal carcinoma, MDS and AML [88, 89]. R-2-HG appears to have an inhibitory effect on a number of histone demethylases including members of the Jumonji transcription factor family (JMJD2A, JMJD2C and JHDM1A/FBXL11), which may contribute to tumorigenesis (Fig. 4) [76]. Furthermore, evidence of hypermethylation of the H3 family of histones H3K4, H3K9, H3K27, H3K36 and H3K79 has been found following mutant IDH1 expression or R-2-HG exposure in multiple human cancer cell lines as well as in normal astrocytes and adipocyte precursors [71, 90, 91]. Lu et al (2012) demonstrated hypermethylation of histone H3K9 in 3T3 fibroblast cells that were exposed to R-2-HG, and this was accompanied by reduced differentiation into mature adipocytes [90]. In the same study they showed immortalized astrocytes transfected with the IDH1 mutation had increased levels of histone methylation. Notably, the particular sites of histone methylation overlapped with those found in IDH1 mutant glioma cells.
Conversely, histone demethylases may also promote cancer formation. Overexpression of JHDM2A has been associated with poor prognosis in colorectal cancer [92], while overexpression of JMJD2C has been demonstrated in esophageal cancer [93], MALT-lymphoma [94] and breast cancer [95]. Furthermore, the oncogenic and oncosuppressive effects of particular histone demethylases depend upon the cell type in which these enzymes are expressed or inhibited [87]. Interestingly, IDH1 wild type gliomas also show evidence of histone hypermethylation. As previously discussed, H3K9 hypermethylation occurs in IDH1 mutated gliomas, but it has also been found in their wild type counterparts [90]. Trimethylation of H3K9 has been strongly linked to IDH1 mutations in oligodendrogliomas and grade II astrocytomas, but has not been associated with IDH1 mutations in grade III/IV astrocytomas, despite the majority of these tumors exhibiting evidence of the hypermethylation phenotype [96]. It may be the case that histone hypermethylation is a common feature broadly across all gliomas rather than being a mechanism by which IDH1 exerts its tumorigenic effects. Alternatively, histone hypermethylation may be propagated by IDH1 mutations in some glioma subtypes (e.g. oligodendrogliomas) but via different mechanisms in others (grade III/IV astrocytomas).
Therapies targeting the IDH1 mutant protein
Small molecule inhibitors targeting the IDH1 mutant protein
Small molecule inhibitors represent a viable strategy for targeting oncogenic enzymes, demonstrated initially by the development of imatinib, a compound inhibiting the bcr-abl fusion protein in hematopoietic malignancies [97]. The first small molecule inhibitor of the mutated IDH1 protein was reported by Popovici-Muller et al (2012) who performed a high-throughput screening of compounds against the R132H IDH1 mutant protein homodimer [98]. Further refining potential candidates with a-KG and NADPH assays, the authors identified a molecule, compound 35, which demonstrated potent inhibition of R-2-HG production in R132H U87 GBM cells and R132C HT1080 chondrosarcoma cells. IC50 values against the two mutant isoforms were less than 0.5 μM in both cell lines while the IC50 for the wild type IDH1 protein was over 20 μM. Additionally, intraperitoneal administration of compound 35 into U87 tumor xenograft mouse models yielded improved IC50 values of 0.07 μM against tumor R-2-HG production. To date, no further studies of compound 35 have been reported by the original authors or other groups. However, given mounting evidence that the mutant IDH1 protein acts as a heterodimer with the wild type protein, this study's approach to screen against a mutant protein homodimer was not ideal.
Recently, a quantitative high throughput compound screen identified ML309 as a potent inhibitor of the R132H mutant IDH1 enzyme [99]. The drug acts a competitive inhibitor of the mutant IDH1, competing with a-KG for the enzymatic active site. As such, drug treatment in GBM cell lines yielded significantly lower levels of R-2-HG in a dose-dependent manner. Additionally, ML309 demonstrated preferential activity against the mutant IDH1 over the wild type, with an IC50 of 96 nM for the former and 35 μM for the latter, respectively. More recently, ML309 was shown to inhibit the R132C IDH1 mutation with similar efficacy [100]. Furthermore, ML309 exhibited good aqueous solubility, was stable in human plasma, and had a moderate half-life of 3.76 hours. Notably, ML309 administration in healthy mice showed absence of BBB penetration. No studies have examined the efficacy of ML309 in a GBM xenograft model where BBB disruption by the tumor would theoretically permit accumulation of the drug in areas of dense tumor.
Similarly, via high throughput screening, another compound, AGI-5198, has been identified as a potent inhibitor of R132H mutated IDH1 [101]. AGI-5198 exhibited higher selectivity than ML309 against mutant IDH1 with an IC50 of 70 nM and an IC50 of >100 μM for the wild type enzyme and may be administered orally. AGI-5198 administration was able to reduce R-2-HG levels in a dose-dependent manner in R132H-mutated TS603 grade III glioma cells and effectively prevented colony formation. Importantly, the drug did not do the same for wild type IDH1 expressing glioma cell lines, further supporting the selectivity of AGI-5198. In addition, in support of an association of IDH1 mutations with the hypermethylation phenotype, ex vivo treatment of TS603 glioma cells with AGI-5198 induced differentiation of nestin-positive neural progenitor cells into glial fibrillary acidic protein (GFAP) and aquaporin-4 (AQ-4)-positive astrocytes with a concomitant reduction in histone methylation associated with these latter genes. Oral administration of AGI-5198 in mice with xenografted subcutaneous tumors also significantly reduced intratumoral R-2-HG levels, diminished immunohistochemical staining of histone methylation, and increased expression of astroglial differentiation genes. Further development of AGI-5198 has led to development of AG-120 and AG-221 (Agios Pharmaceuticals, Cambridge, MA), orally administered drugs targeting IDH1 and IDH2 mutations, respectively. As such, a multicentre, open-label, dose escalation phase 1 clinical trial was started in March 2014, studying the safety and tolerability of AG-120, in patients with advanced hematologic malignancies and advanced solid tumors. Likewise, a phase 1 trial for AG-221 was launched in September 2013 for advanced hematologic cancers. Preliminary results from the AG-221 phase 1 trial have demonstrated good patient tolerance with no dose-limiting toxicities. Reportedly, 14/25 patients responded objectively to treatment and 6 patients experienced complete remissions (Press release by Agios Pharmaceuticals, dated April 6, 2014; accessed at http://investor.agios.com/phoenix.zhtml?c=251862&p=irol-newsArticle&ID=1916041). In addition, AG-221 treatment has correlated with reductions in plasma R-2-HG levels. Similar early results are highly anticipated for the AG-120 trial.
Another group recently identified a small molecule inhibitor of R132H IDH1 from a screen of a commercially available library of three million compounds (Exelixis, Cambridge, MA) [48]. The compound, EXEL-9324, was found to be the most potent inhibitor of R-2-HG production and exhibited an IC50 of 800 nM against the a-KG to R-2-HG reaction catalysed by the R132H/wild type heterodimer IDH1 protein, transfected into E. Coli cells. Importantly, the authors also demonstrated that EXEL-9324 selectively targeted this oncogenic heterodimer complex as the affinities of the compound for the wild type and mutant homodimers were exceedingly diminished. Furthermore, this study confirmed previous theories that the mutant IDH1 protein depends upon the presence of a wild type IDH1 protein for production of the metabolite, R-2-HG. As such, additional work studying the in vivo efficacy of EXEL-9324 may potentially contribute greatly to IDH1 targeted therapies in the future.
Instead of directly inhibiting the mutant IDH1 protein, additional compounds have been identified that similarly result in decreased R-2-HG production. One such example is zaprinast, a phosphodiesterase-5 inhibitor (PDE5), which was identified via a high throughput fluorimetric assay for R-2-HG [102]. Zaprinast mediates its anti-2-HG activity via non-competitive inhibition of glutaminase, which converts glutamine to glutamate, the latter being the precursor for a-KG and subsequently, R-2-HG. Administration of this drug in R132H IDH1 mutated immortalized human astrocytes as well as in R132C IDH1 mutated HT1080 fibrosarcoma cells led to potent reduction of R-2-HG in a concentration dependent manner. Furthermore, these results were also reproduced in a HT1080 xenograft model. Interestingly, the concentration of zaprinast required to significantly reduce 2-HG levels exceeded that against PDE5 by an approximate magnitude of ten, suggesting anti-2-HG activity may have resulted from off-target effects. It is yet to be seen whether the doses of zaprinast required for therapeutic efficacy lead to in vivo toxicity. Additionally, it is unknown whether zaprinast has any ability to penetrate the BBB. However, a handful of studies have demonstrated that inhibition of glutaminase inhibits glioma cells, suggesting that targeting glutaminase may be a potential strategy for inhibiting mutant IDH1 enzymatic activity [103-105].
Vaccine immunotherapy against the IDH1 mutant protein
Development of glioma-specific vaccine therapies has garnered interest as a way of therapeutically modulating the native immune system to recognize and destroy tumor cells. To date, none of the phase 1 or 2 clinical trials of vaccine immunotherapies have specifically sought to target the IDH1 mutated epitope. Furthermore, in their phase I/IIa trial of an autologous formalin-fixed tumor vaccine for newly diagnosed GBM (administered with fractionated radiotherapy and temozolomide), Ishikawa and colleagues did not find any significant association between vaccine response (induction of delayed-type hypersensitivity) and IDH1 R132H mutation status [106].
Recently, however, a group published their pre-clinical work in development of a vaccine immunotherapy targeting the IDH1 mutant protein [107]. Using 15-mer peptides from the R132H IDH1 mutant protein loaded onto MHC class II complexes, vaccination of MHC-humanized transgenic mice generated robust Th1-cell responses as evidenced by increased interferon-gamma production and detectable levels of anti-IDH1 (R132H) in the serum. Notably, these effects were not seen with homologous peptides from the IDH1 wild type protein. Furthermore, these findings were reproduced in IDH1 (R132H) mutated sarcomas in mouse xenografts, resulting in potent tumor growth suppression and absence of overt toxicities. Interestingly, the authors screened 25 patients with R132H IDH1 mutated gliomas and found detectable levels of IFN-gamma producing Th1 cells against this specific epitope in four patients. However, it is unclear whether the presence of an anti-IDH1 mutant T-cell response in these select patients conferred any survival benefit. HLA typing of all 25 patients was non-specific suggesting the mutant IDH1 protein is not limited to any particular HLA class II type. Taken together, although only a single study, there will likely be increased efforts to develop novel immunotherapies that target the IDH1 mutant protein. It is yet to be seen whether the results of this study are reproducible in tumors protected by the BBB. Furthermore, many questions remain regarding the prognostic significance of patients who are able to mount an IDH1 mutant specific immune response without intervention.
Conclusions
Just six years since the IDH1 mutation was first discovered in GBM, our understanding of the prevalence and pathogenesis of this mutation in both CNS and non-CNS tumors has grown at a rapid rate. It is well established that IDH1 is an important mutation in LGG and secondary GBM, and this knowledge is being readily applied to patient care. Classifications based upon IDH1 mutation status are increasingly being used in clinical practice [7]. Diagnosis of IDH1 mutations has been able to provide important diagnostic and prognostic information for patients, removing some of the burden of uncertainty. Furthermore, as previously mentioned, two Phase-1 multicentre dose escalation trials are underway that are evaluating an oral medication targeting the IDH1 mutant protein in patients with hematological and solid malignancies. As more precise molecular targets become elucidated, the hope is that this will provide a much-needed boost for treatment options.
As demonstrated in this review, multiple potential mechanisms for the role of IDH1 in tumorigenesis have been proposed. The majority of research has focused on the role of R-2-HG-mediated effects on a number of key cellular processes. This has revealed that R-2-HG has a diverse set of targets that in theory, could explain how IDH1 mutations mediate tumor formation. The data described in this review have thus far revealed, unsurprisingly, that a single simple explanation is unlikely. Both DNA and histone methylation generate epigenetic changes resulting in altered cellular developmental programs that may be unique to IDH1 mutated tumors. It is also likely that HIFs play some role, although current evidence suggests that this may be counter-intuitive to what we understand of HIF functioning in other cancers. Much of the research has focused on the a-KG-dependent dioxygenase TET2, but it should be noted that there are over 70 a-KG-dependent dioxygenases that could be potentially involved in the oncogenesis of IDH1 mutated malignancies. To truly understand the effects of the IDH1 mutation, additional research is needed to cover the whole spectrum of targets, rather than reliance on previously investigated mechanisms. Although many additional questions remain, research in the oncogenic mechanisms of the IDH1 mutation has provided rich new findings for the field of cancer biology. With continued research efforts, it is likely these questions may soon be answered.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health (NIH).
Abbreviations
GBM: Glioblastoma; TCGA: The Cancer Genome Atlas; IDH1: Isocitrate dehydrogenase; CNS: Central nervous system; LOH: Loss of heterozygosity; 2-HG: 2-hydroxyglutarate; AML: Acute myeloid leukemia; MDS: Myelodysplastic syndrome; MPN: Myeloproliferative neoplasms; a-KG: Alpha-ketoglutarate; TCA: Tricarboxylic acid cycle; R-2-HG: R-2-hydroxyglutarate; EPO: Erythropoietin; PHD: Prolyl hydroxylases; HIFs: Hypoxia-inducible factors; VHL: von-Hippel Lindau; NF-κB: nuclear factor-κB; PLOD: Procollagen-lysine, 2-oxoglutarate 5-dioxygenase; BBB: Blood brain barrier; ER: Endoplasmic reticulum; LGG: Low-grade gliomas; G-CIMP: Glioma-CpG island methylator phenotype; TET: Tet methylcytosine dioxygenase; 5mc: 5-methylcytosine; 5hmc: 5-hydroxymethylcytosine; CMML: Chronic myelomonocytic leukemia; GFAP: Glial fibrillary acidic protein; AQ-4: Aquaporin-4; PDE5: Phosphodiesterase-5.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-oncology. 2012;14(Suppl 5):v1-49 doi:10.1093/neuonc/nos218
2. Walker MD, Alexander E Jr, Hunt WE, MacCarty CS, Mahaley MS Jr, Mealey J Jr. et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. Journal of neurosurgery. 1978;49:333-43 doi:10.3171/jns.1978.49.3.0333
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987-96 doi:10.1056/NEJMoa043330
4. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8 doi:10.1038/nature07385
5. Frezza C, Gottlieb E. Mitochondria in cancer: not just innocent bystanders. Seminars in cancer biology. 2009;19:4-11 doi:10.1016/j.semcancer.2008.11.008
6. Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. The American journal of pathology. 2007;170:1445-53 doi:10.2353/ajpath.2007.070011
7. Dunn GP, Andronesi OC, Cahill DP. From genomics to the clinic: biological and translational insights of mutant IDH1/2 in glioma. Neurosurgical focus. 2013;34:E2. doi:10.3171/2012.12.FOCUS12355
8. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta neuropathologica. 2008;116:597-602 doi:10.1007/s00401-008-0455-2
9. SongTao Q, Lei Y, Si G, YanQing D, HuiXia H, XueLin Z. et al. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer science. 2012;103:269-73 doi:10.1111/j.1349-7006.2011.02134.x
10. Beiko J, Suki D, Hess KR, Fox BD, Cheung V, Cabral M. et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro-oncology. 2014;16:81-91 doi:10.1093/neuonc/not159
11. Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL. et al. Genetic pathways to glioblastoma: a population-based study. Cancer research. 2004;64:6892-9 doi:10.1158/0008-5472.CAN-04-1337
12. Fujisawa H, Reis RM, Nakamura M, Colella S, Yonekawa Y, Kleihues P. et al. Loss of heterozygosity on chromosome 10 is more extensive in primary (de novo) than in secondary glioblastomas. Laboratory investigation; a journal of technical methods and pathology. 2000;80:65-72
13. Nakamura M, Yang F, Fujisawa H, Yonekawa Y, Kleihues P, Ohgaki H. Loss of heterozygosity on chromosome 19 in secondary glioblastomas. Journal of neuropathology and experimental neurology. 2000;59:539-43
14. Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM. et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta neuropathologica. 2012;124:615-25 doi:10.1007/s00401-012-1031-3
15. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A. et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta neuropathologica. 2009;118:469-74 doi:10.1007/s00401-009-0561-9
16. Pusch S, Schweizer L, Beck AC, Lehmler JM, Weissert S, Balss J. et al. D-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta neuropathologica communications. 2014;2:19. doi:10.1186/2051-5960-2-19
17. Ward PS, Lu C, Cross JR, Abdel-Wahab O, Levine RL, Schwartz GK. et al. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. The Journal of biological chemistry. 2013;288:3804-15 doi:10.1074/jbc.M112.435495
18. Molenaar RJ, Radivoyevitch T, Maciejewski JP, van Noorden CJ, Bleeker FE. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochimica et biophysica acta. 2014;1846:326-41 doi:10.1016/j.bbcan.2014.05.004
19. Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE. et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer research. 2009;69:9065-72 doi:10.1158/0008-5472.CAN-09-2307
20. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF. et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709-22
21. Kros JM. From expert opinion to evidence-based: changes in the gold standard of primary brain tumour diagnosis. The Journal of pathology. 2007;213:1-3 doi:10.1002/path.2201
22. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. The New England journal of medicine. 2009;361:1058-66 doi:10.1056/NEJMoa0903840
23. Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D. et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:2348-55 doi:10.1200/JCO.2009.27.3730
24. Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L. et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:3636-43 doi:10.1200/JCO.2010.28.3762
25. Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A. et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116:2122-6 doi:10.1182/blood-2009-11-250878
26. Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B. et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24:1094-6 doi:10.1038/leu.2010.52
27. Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C. et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621-5 doi:10.1126/science.1231677
28. Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F. et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. The Journal of pathology. 2011;224:334-43 doi:10.1002/path.2913
29. Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F. et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nature genetics. 2011;43:1262-5 doi:10.1038/ng.994
30. Rawlings CE 3rd, Bullard DE, Burger PC, Friedman AH. A case of Ollier's disease associated with two intracranial gliomas. Neurosurgery. 1987;21:400-3
31. Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR. et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091-100 doi:10.1038/onc.2012.315
32. Horbinski C, Kofler J, Yeaney G, Camelo-Piragua S, Venneti S, Louis DN. et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain pathology. 2011;21:564-74 doi:10.1111/j.1750-3639.2011.00480.x
33. Hayden JT, Fruhwald MC, Hasselblatt M, Ellison DW, Bailey S, Clifford SC. Frequent IDH1 mutations in supratentorial primitive neuroectodermal tumors (sPNET) of adults but not children. Cell cycle. 2009;8:1806-7
34. Dalziel K. Isocitrate dehydrogenase and related oxidative decarboxylases. FEBS letters. 1980;117(Suppl):K45-55
35. Geisbrecht BV, Liang X, Morrell JC, Schulz H, Gould SJ. The mouse gene PDCR encodes a peroxisomal delta(2), delta(4)-dienoyl-CoA reductase. The Journal of biological chemistry. 1999;274:25814-20
36. Kim SY, Lee SM, Tak JK, Choi KS, Kwon TK, Park JW. Regulation of singlet oxygen-induced apoptosis by cytosolic NADP+-dependent isocitrate dehydrogenase. Molecular and cellular biochemistry. 2007;302:27-34 doi:10.1007/s11010-007-9421-x
37. van Roermund CW, Hettema EH, Kal AJ, van den Berg M, Tabak HF, Wanders RJ. Peroxisomal beta-oxidation of polyunsaturated fatty acids in Saccharomyces cerevisiae: isocitrate dehydrogenase provides NADPH for reduction of double bonds at even positions. The EMBO journal. 1998;17:677-87 doi:10.1093/emboj/17.3.677
38. Koh HJ, Lee SM, Son BG, Lee SH, Ryoo ZY, Chang KT. et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. The Journal of biological chemistry. 2004;279:39968-74 doi:10.1074/jbc.M402260200
39. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K. et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380-4 doi:10.1038/nature10602
40. Ronnebaum SM, Ilkayeva O, Burgess SC, Joseph JW, Lu D, Stevens RD. et al. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. The Journal of biological chemistry. 2006;281:30593-602 doi:10.1074/jbc.M511908200
41. Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F. et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:4482-90 doi:10.1200/JCO.2010.33.8715
42. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739-44 doi:10.1038/nature08617
43. Pietrak B, Zhao H, Qi H, Quinn C, Gao E, Boyer JG. et al. A tale of two subunits: how the neomorphic R132H IDH1 mutation enhances production of alphaHG. Biochemistry. 2011;50:4804-12 doi:10.1021/bi200499m
44. Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. Journal of inherited metabolic disease. 2012;35:571-87 doi:10.1007/s10545-012-9462-5
45. Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M. L-2-hydroxyglutaric aciduria, a defect of metabolite repair. Journal of inherited metabolic disease. 2007;30:681-9 doi:10.1007/s10545-007-0487-0
46. Struys EA, Verhoeven NM, Ten Brink HJ, Wickenhagen WV, Gibson KM, Jakobs C. Kinetic characterization of human hydroxyacid-oxoacid transhydrogenase: relevance to D-2-hydroxyglutaric and gamma-hydroxybutyric acidurias. Journal of inherited metabolic disease. 2005;28:921-30 doi:10.1007/s10545-005-0114-x
47. Jin G, Reitman ZJ, Duncan CG, Spasojevic I, Gooden DM, Rasheed BA. et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer research. 2013;73:496-501 doi:10.1158/0008-5472.CAN-12-2852
48. Brooks E, Wu X, Hanel A, Nguyen S, Wang J, Zhang JH. et al. Identification and Characterization of Small-Molecule Inhibitors of the R132H/R132H Mutant Isocitrate Dehydrogenase 1 Homodimer and R132H/Wild-Type Heterodimer. Journal of biomolecular screening. 2014;19:1193-200 doi:10.1177/1087057114541148
49. Garber K. Oncometabolite? IDH1 discoveries raise possibility of new metabolism targets in brain cancers and leukemia. Journal of the National Cancer Institute. 2010;102:926-8 doi:10.1093/jnci/djq262
50. Patay Z, Mills JC, Lobel U, Lambert A, Sablauer A, Ellison DW. Cerebral neoplasms in L-2 hydroxyglutaric aciduria: 3 new cases and meta-analysis of literature data. AJNR American journal of neuroradiology. 2012;33:940-3 doi:10.3174/ajnr.A2869
51. Kloosterhof NK, Bralten LB, Dubbink HJ, French PJ, van den Bent MJ. Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? The lancet oncology. 2011;12:83-91 doi:10.1016/S1470-2045(10)70053-X
52. Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nature chemical biology. 2008;4:152-6 doi:10.1038/nchembio0308-152
53. Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology. 2004;19:176-82 doi:10.1152/physiol.00001.2004
54. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P. et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261-5 doi:10.1126/science.1170944
55. Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II. et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes & development. 2012;26:2038-49 doi:10.1101/gad.198200.112
56. Wang G, Sai K, Gong F, Yang Q, Chen F, Lin J. Mutation of isocitrate dehydrogenase 1 induces glioma cell proliferation via nuclear factor-kappaB activation in a hypoxia-inducible factor 1-alpha dependent manner. Molecular medicine reports. 2014;9:1799-805 doi:10.3892/mmr.2014.2052
57. Qin N, de Cubas AA, Garcia-Martin R, Richter S, Peitzsch M, Menschikowski M. et al. Opposing effects of HIF1alpha and HIF2alpha on chromaffin cell phenotypic features and tumor cell proliferation: Insights from MYC-associated factor X. International journal of cancer Journal international du cancer. 2014;135:2054-64 doi:10.1002/ijc.28868
58. Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL. et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Molecular and cellular biology. 2005;25:5675-86 doi:10.1128/MCB.25.13.5675-5686.2005
59. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S. et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484-8 doi:10.1038/nature10898
60. Williams SC, Karajannis MA, Chiriboga L, Golfinos JG, von Deimling A, Zagzag D. R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1alpha upregulation in adult glioma. Acta neuropathologica. 2011;121:279-81 doi:10.1007/s00401-010-0790-y
61. Acker T, Diez-Juan A, Aragones J, Tjwa M, Brusselmans K, Moons L. et al. Genetic evidence for a tumor suppressor role of HIF-2alpha. Cancer cell. 2005;8:131-41 doi:10.1016/j.ccr.2005.07.003
62. Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends in genetics: TIG. 2004;20:33-43 doi:10.1016/j.tig.2003.11.004
63. Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619-28 doi:10.1242/dev.01037
64. Rautavuoma K, Takaluoma K, Sormunen R, Myllyharju J, Kivirikko KI, Soininen R. Premature aggregation of type IV collagen and early lethality in lysyl hydroxylase 3 null mice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:14120-5 doi:10.1073/pnas.0404966101
65. Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N. et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nature genetics. 2013;45:923-6 doi:10.1038/ng.2668
66. Suzuki H, Yamamoto E, Maruyama R, Niinuma T, Kai M. Biological significance of the CpG island methylator phenotype. Biochemical and biophysical research communications. 2014 doi:10.1016/j.bbrc.2014.07.007
67. Zhang L, Wang M, Wang W, Mo J. Incidence and prognostic value of multiple gene promoter methylations in gliomas. Journal of neuro-oncology. 2014;116:349-56 doi:10.1007/s11060-013-1301-5
68. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010;17:510-22 doi:10.1016/j.ccr.2010.03.017
69. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18:553-67 doi:10.1016/j.ccr.2010.11.015
70. Li S, Chowdhury R, Liu F, Chou AP, Li T, Mody RR. et al. Tumor suppressive miR-148a is silenced by CpG island hypermethylation in IDH1 mutant gliomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014 doi:10.1158/1078-0432.CCR-14-0234
71. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479-83 doi:10.1038/nature10866
72. Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS. et al. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer research. 2001;61:4956-60
73. Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A. et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656-9 doi:10.1038/nature11323
74. Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C. et al. IDH2 mutation induced histone and DNA hypermethylation is progressively reversed by small molecule inhibition. Blood. 2014 doi:10.1182/blood-2013-10-533604
75. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-5 doi:10.1126/science.1170116
76. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR. et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO reports. 2011;12:463-9 doi:10.1038/embor.2011.43
77. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19:17-30 doi:10.1016/j.ccr.2010.12.014
78. Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M. et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144-7 doi:10.1182/blood-2009-03-210039
79. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS. et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839-43 doi:10.1038/nature09586
80. Perez C, Martinez-Calle N, Martin-Subero JI, Segura V, Delabesse E, Fernandez-Mercado M. et al. TET2 mutations are associated with specific 5-methylcytosine and 5-hydroxymethylcytosine profiles in patients with chronic myelomonocytic leukemia. PloS one. 2012;7:e31605. doi:10.1371/journal.pone.0031605
81. Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L. et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics: official journal of the DNA Methylation Society. 2012;7:201-7 doi:10.4161/epi.7.2.19015
82. Kim YH, Pierscianek D, Mittelbronn M, Vital A, Mariani L, Hasselblatt M. et al. TET2 promoter methylation in low-grade diffuse gliomas lacking IDH1/2 mutations. Journal of clinical pathology. 2011;64:850-2 doi:10.1136/jclinpath-2011-200133
83. Muller T, Gessi M, Waha A, Isselstein LJ, Luxen D, Freihoff D. et al. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. The American journal of pathology. 2012;181:675-83 doi:10.1016/j.ajpath.2012.04.017
84. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12-27 doi:10.1016/j.cell.2012.06.013
85. Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407-12 doi:10.1038/nature05915
86. Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nature reviews Cancer. 2010;10:457-69 doi:10.1038/nrc2876
87. Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes & development. 2008;22:1115-40 doi:10.1101/gad.1652908
88. Hu Z, Gomes I, Horrigan SK, Kravarusic J, Mar B, Arbieva Z. et al. A novel nuclear protein, 5qNCA (LOC51780) is a candidate for the myeloid leukemia tumor suppressor gene on chromosome 5 band q31. Oncogene. 2001;20:6946-54 doi:10.1038/sj.onc.1204850
89. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A. et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360-3 doi:10.1038/nature08672
90. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474-8 doi:10.1038/nature10860
91. Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR. et al. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome research. 2012;22:2339-55 doi:10.1101/gr.132738.111
92. Uemura M, Yamamoto H, Takemasa I, Mimori K, Hemmi H, Mizushima T. et al. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:4636-46 doi:10.1158/1078-0432.CCR-10-0407
93. Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M. et al. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer research. 2000;60:4735-9
94. Vinatzer U, Gollinger M, Mullauer L, Raderer M, Chott A, Streubel B. Mucosa-associated lymphoid tissue lymphoma: novel translocations including rearrangements of ODZ2, JMJD2C, and CNN3. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:6426-31 doi:10.1158/1078-0432.CCR-08-0702
95. Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP. et al. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. 2009;28:4491-500 doi:10.1038/onc.2009.297
96. Venneti S, Felicella MM, Coyne T, Phillips JJ, Gorovets D, Huse JT. et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. Journal of neuropathology and experimental neurology. 2013;72:298-306 doi:10.1097/NEN.0b013e3182898113
97. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S. et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine. 1996;2:561-6
98. Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F. et al. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS medicinal chemistry letters. 2012;3:850-5 doi:10.1021/ml300225h
99. Davis M, Pragani R, Popovici-Muller J, Gross S, Thorne N, Salituro F. et al. ML309: A potent inhibitor of R132H mutant IDH1 capable of reducing 2-hydroxyglutarate production in U87 MG glioblastoma cells. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD). 2010
100. Davis MI, Gross S, Shen M, Straley KS, Pragani R, Lea WA. et al. Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. The Journal of biological chemistry. 2014;289:13717-25 doi:10.1074/jbc.M113.511030
101. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C. et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626-30 doi:10.1126/science.1236062
102. Elhammali A, Ippolito JE, Collins L, Crowley J, Marasa J, Piwnica-Worms D. A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies Zaprinast as a glutaminase inhibitor. Cancer discovery. 2014;4:828-39 doi:10.1158/2159-8290.CD-13-0572
103. Martin-Rufian M, Nascimento-Gomes R, Higuero A, Crisma AR, Campos-Sandoval JA, Gomez-Garcia MC. et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. Journal of molecular medicine. 2014;92:277-90 doi:10.1007/s00109-013-1105-2
104. Szeliga M, Bogacinska-Karas M, Rozycka A, Hilgier W, Marquez J, Albrecht J. Silencing of GLS and overexpression of GLS2 genes cooperate in decreasing the proliferation and viability of glioblastoma cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:1855-62 doi:10.1007/s13277-013-1247-4
105. Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV. et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer research. 2010;70:8981-7 doi:10.1158/0008-5472.CAN-10-1666
106. Ishikawa E, Muragaki Y, Yamamoto T, Maruyama T, Tsuboi K, Ikuta S. et al. Phase I/IIa trial of fractionated radiotherapy, temozolomide, and autologous formalin-fixed tumor vaccine for newly diagnosed glioblastoma. Journal of neurosurgery. 2014;121:543-53 doi:10.3171/2014.5.JNS132392
107. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J. et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324-7 doi:10.1038/nature13387
Author contact
![]() Corresponding authors: Zhengping Zhuang, M.D., Ph.D. P: (301) 435-8445; F: (301) 402-0536; E: zhuangpnih.gov. John D. Heiss, M.D. P: (301) 594-8112; F: (301) 402-0380; E: heissjnih.gov
Corresponding authors: Zhengping Zhuang, M.D., Ph.D. P: (301) 435-8445; F: (301) 402-0536; E: zhuangpnih.gov. John D. Heiss, M.D. P: (301) 594-8112; F: (301) 402-0380; E: heissjnih.gov