Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(1):48-56. doi:10.7150/ijms.10035 This issue Cite
Research Paper
Astrocytes Protect Neurons from Aβ1-42 Peptide-Induced Neurotoxicity Increasing TFAM and PGC-1 and Decreasing PPAR-γ and SIRT-1
Diana Aguirre-Rueda, Sol Guerra-Ojeda, Martin Aldasoro, Antonio Iradi, Elena Obrador, Angel Ortega, M. Dolores Mauricio, Jose Mª Vila, Soraya L. Valles ![]()
Department of Physiology. School of Medicine, University of Valencia. Spain.
Received 2014-7-3; Accepted 2014-10-21; Published 2015-1-1
Abstract
One of the earliest neuropathological events in Alzheimer's disease is accumulation of astrocytes at sites of Aβ1-42 depositions. Our results indicate that Aβ1-42 toxic peptide increases lipid peroxidation, apoptosis and cell death in neurons but not in astrocytes in primary culture. Aβ1-42-induced deleterious neuronal effects are not present when neurons and astrocytes are mixed cultured. Stimulation of astrocytes with toxic Aβ1-42 peptide increased p-65 and decreased IκB resulting in inflammatory process. In astrocytes Aβ1-42 decreases protein expressions of sirtuin 1 (SIRT-1) and peroxisome proliferator-activated receptor γ (PPAR-γ) and over-expresses peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1) and mitochondrial transcription factor A (TFAM), protecting mitochondria against Aβ1-42-induced damage and promoting mitochondrial biogenesis.
In summary our data suggest that astrocytes may have a key role in protecting neurons, increasing neural viability and mitochondrial biogenesis, acquiring better oxidative stress protection and perhaps modulating inflammatory processes against Aβ1-42 toxic peptide. This might be a sign of a complex epigenetic process in Alzheimer's disease development.
Keywords: Alzheimer's Disease, MnSOD, PPAR-γ, TFAM, PGC-1, NF-κB.
Introduction
The metabolic processing of the amyloid precursor protein (APP) produces mainly two peptide fractions of amyloid β peptide (Aβ), Aβ1-40 and Aβ1-42. The accumulation and precipitation of Aβ1-42 peptide has been suggested as a neuropathological event associated with the progress and development of Alzheimer's disease (AD) [1, 2]. Extracellular Aβ1-42 deposits can cause oxidative stress, inflammation and mitochondrial dysfunction [3, 4, 5]. It has been shown in vitro and in vivo studies that the Aβ1-42 enters the mitochondria causing an increase in reactive oxygen species, interruption of the electron transport chain and blockade of the ATP production [6, 7]. A deficiency in the function of the mitochondrial respiratory chain could affect the survival of cells associated with AD resulting in adverse impact for long-term health [8]. Moreover extracellular Aβ1-42 peptide induces oxidative stress and causes damage to mitochondrial DNA (mtDNA) which encodes the 13 key subunits of the respiratory chain, essential for respiratory function, susceptible to oxidative damage [9,10]. The mtDNA maintenance, including nucleotide formation as well as mtDNA stabilization and transcription is in charge of mitochondrial transcription factor A (TFAM) [11]. Recent studies have indicated that TFAM and PGC-1 over-expression may inhibit mitochondrial ROS generation and improve mitochondrial respiratory function [12, 13]. Furthermore over-expression of TFAM has been shown protect mitochondria against Aβ-induced oxidative damage in neurons [14].
Glial cells interact with Aβ1-42 and support neurons, but whether astrocytes could participate in regulating the Aβ-induced brain damage is, at the moment, poorly known. Here we evaluated the role of astrocytes in the neuro-toxic effects of Aβ peptides. For this purpose we used primary astrocytes, neurons and mixed-cultures in the absence and in the presence of toxic Aβ1-42 peptide or non-toxic Aβ40-1 (control) peptide. Our results demonstrate that Aβ1-42 (5 µM) induces apoptotic cell death in neurons. However in mixed-cultures, astrocytes protect neurons against Aβ1-42-induced toxicity. Mechanistically astrocytes-induced neuron protection involves over-expression of TFAM and PGC-1, and decrease in PPAR-γ and SIRT-1.
Experimental Procedures
Materials
3-(4,5-dimethyl-2-thiazolyl)-2,5-dipheniyl-2H-tetrazolium bromide (MTT) was obtained from Sigma Chemical Co. (St Louis, MO). Dulbecco's modified Eagle's medium (DMEM) and foetal bovine serum (FBS) were obtained from Gibco (Gibco Invitrogen Corporation, Barcelona, Spain). Western Blot Chemiluminescent Detection System (ECL) was from Amersham (Amersham Biosciences, Barcelona, Spain). Antibodies: monoclonal anti-cytochrome c antibody (anti-Cytochrome c) (1:500), polyclonal anti-manganese superoxide dismutase (anti-MnSOD) (1:250), monoclonal anti-p65 (1:250), monoclonal anti- PGC-1 (1:250), monoclonal anti-TFAM (1:250), monoclonal anti-IκB (1:250), polyclonal anti-PPAR-γ (1:300), monoclonal anti-SIRT-1 (1:250), and monoclonal anti-tubuline (1:3000) antibodies (Sigma Aldrich, Madrid, Spain) were used. All other reagents were of analytical or culture grade purity. The oligomers Aβ (40-1 and 1-42), were prepared following manufacture instructions such as indicates Sigma-Aldrich biotechnology. Briefly, the peptides were dissolved in H2O and for assembly the oligomer preparations were heated for 24 h at 37ºC.
Primary culture of cortical astrocytes
All animals were handled according to the rules established by the bioethics committee of the School of Medicine, University of Valencia, Spain. Cerebral cortical astrocytes were isolated from rat fetuses of 21 days gestation. Fetuses were obtained by cesarean section and decapitated. Cerebral cortices were removed and cut into 1mm cubes and triturated 10-15 times through a Pasteur pipette. After centrifugation at 1000 rpm for 5 min the pellet was resuspended in DMEM containing 20% fetal bovine serum (FBS), supplemented with L-glutamine (1%), HEPES (10 mM), fungizone (1%), and antibiotics (1%). Cells were plated on T75 culture flask pretreated with poli-L-lysine. Cultures were maintained in a humidified atmosphere of 5% CO2/95% air at 37°C and allowed to grow to confluence and used at 15-20 days in vitro. After 1week of culture, the FBS content was reduced to 10%, and the medium was changed twice a week. The purity of astrocytes was assessed by immunofluorescence using anti-glial fibrillary acidic protein (anti-GFAP, astrocyte marker: Sigma-Aldrich, Madrid, Spain), anti-CD-68 (microglial marker: Serotec, Kidlington, UK), anti-myelin basic protein (oligodendroglial marker; Sigma-Aldrich, Madrid, Spain) and anti- microtubule-associated protein 2 (anti-MAP2, neuronal marker; Sigma-Aldrich, Madrid, Spain). The astrocytes were found to be at least 99% glial fibrillary acidic protein positive. No cells were found to express CD-68, myelin basic protein or microtubule-associated protein-2. For all of the experiments we used toxin-free sterile culture materials.
Neurons and neurons-astrocytes mixed cultures
Cerebral cortical neurons were isolated from rat fetuses of 14 or 15 days gestation. Briefly, cerebral cortices were removed and dissociated mechanically, by pipetting 10 times with 10ml DMEM. The cell suspension was filtered through nylon mesh with a pore size of 90 μm. Cell suspension was plated on T75 culture flask pretreated with poli-L-lysine. After attachment of the cells, the culture medium was changed to DMEM containing 10% FBS supplemented with antibiotics (1%) and fungizone (1%). Cultures were grown in a humidified atmosphere of 5% CO2/95% air at 37ºC. After 3 days, cells were exposed to 10 μM cytosine β-D arabino-furanoside for 24 h to inhibit proliferation of non-neuronal cells. The purity of neurons and possible contamination by astrocytes were assessed by immunofluorescence using antibodies anti-GFAP and anti-MAP-2. Under these conditions, 99% of the cells were neurons. Mixed cultures cells were not exposed to cytosine β-D arabino-furanoside allowing the proliferation of non-neuronal cells. Ten days after seeding approximately 60% of cells were astrocytes and 40% neurons. At this time, 5 μM Aβ1-42 (toxic) or Aβ40-1 (control) were added and assays were performed at indicated times. Treatment of cultures with Aβ40-1 did not produce any effects compared with cultures in the absence of any peptide (data not shown).
MTT assay
Cell viability of the cultures was determined by the MTT assay [15]. Neurons and/or astrocytes were plated in 96 well cultures. Aβ40-1 (control) and Aβ1-42 peptides were added to wells during 24h. After cell treatments the medium was removed and the cortical cells were incubated with red free medium and MTT solution [0.5 mg/ml, prepared in phosphate buffer saline (PBS) solution] for 4 h at 37ºC. Finally the medium was removed and formazan particles were dissolved in dimethyl sulfoxide (DMSO). Cell viability, defined as the relative amount of MTT reduction was determined by spectrophotometry at 570 nm.
Caspase-3 activity assay
Caspase activity was performed with the Apo Alert CPP32/caspase 3 assay kit according to the manufacturer's protocol (CLONTECH, Palo Alto, CA). Adherent cells were collected, centrifuged and washed twice with ice-cold PBS and stored as a pellet at -80 ºC. Cell pellets were solubilised in lyses buffer and centrifuged to remove debris and samples containing equal amounts of protein were incubated with 50 μm N-acetyl-Asp-Glut-Val-Asp-AFC (7-amino-4-trifluoromethyl coumarin) 1h at 37ºC. The samples were analyzed using an enzyme-linked immunosorbent assay plate reader with excitation of 360 nm and emission of 530 nm.
Malondialdehyde determination
Malondialdehyde (MDA) levels were assessed by high-performance liquid chromatography. Cells were homogenized and centrifuged, and the supernatants collected and stored on ice. Samples were hydrolyzed by boiling in diluted phosphoric acid. MDA, one of the low- molecular weight end products formed by lipid-peroxidation, reacted with thiobarbituric acid (TBA) to form MDA-TBA adducts which were eluted with methanol-phosphate buffer to be quantified by spectrophotometry at 532 nm.
Western blot analysis
Cultured cells were treated with lysis buffer and then mechanically degraded to release the proteins. Protein concentration was determined using modified Lowry method [16]. Loading buffer (0.125 M Tris-HCl, pH 6.8, 2% SDS, 0.5% (v/v) 2-mercaptoethanol, 1% bromophenolblue and 19% glycerol) was added to protein sample and heated for 5 min at 95ºC. Proteins were separated on SDS-PAGE gels and transferred to nitrocellulose membranes in a humid environment using a transfer buffer (25 mM Tris, 190 mM glycine, 20% methanol). Membranes were blocked with 5% milk in TBS (0.05% Tween-20) and were incubated with primary antibodies overnight at 4ºC. Membranes were washed 3 times with wash buffer TBS-T (TBS, 0.2% Tween-20) and were incubated with a secondary anti-rabbit IgG or anti-mouse IgG (Cell Signalling Technologies Danvers, MA) antibody conjugated to the enzyme horseradish peroxidase (HRP) for 1 h. Membranes were washed three times and proteins were detected using the ECL method as specified by the manufacturer. Autoradiography signals were assessed using digital image system ImageQuant LAS 4000 (GE Healthcare).
Peroxide levels determination
To determine peroxide levels we used a modification of the method described by Barja et al. [16]. Cells were washed twice with PBS and then incubated at 37ºC with a PBS solution containing 0.1 mM homovanilic acid and 6 U/ml horseradish peroxidase. Incubation was stopped at 5 min with cold glycine buffer (2M) containing 50 mM EDTA and 2.2 M NaOH. Supernatant was measured by fluorescence (312 nm excitation and 420 nm emission).
Statistical methods
Values are expressed as mean ± S.D. Differences between groups were assessed by one-way analysis of variance (ANOVA). Statistical significance was accepted at P ≤ 0.05. Data sets in which F was significant were examined by a modified t-test.
Results
Astrocytes protect neurons from Aβ-induced cell death
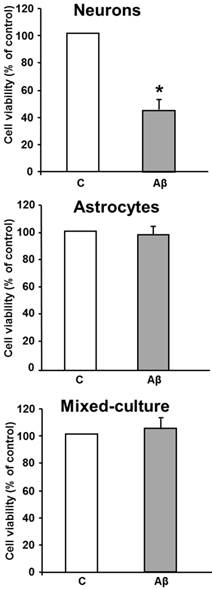
As a first step we evaluated Aβ-induced cell death in cultured neurons and/or astrocytes. As shown in Figure 1, treatment with Aβ1-42 peptide (5 μM 24 hours) caused a decrease in cell viability of about 50% compared with control neurons. However, treatment of astrocytes with the toxic peptide did not induce any significant changes in cell viability compared with control untreated astrocytes. Moreover, when neurons and astrocytes were mixed-cultures we did not detect neuron cell death after toxic peptide addition as compared with controls.
Aβ1-42 induces loss of cell viability in neurons but not in astrocytes and in mixed-culture cells. Cell viability was determined by MTT assay in cells treated for 24 h with 5 μM Aβ40-1 (control, C) or 5 μM Aβ1-42 (Aβ). Data are mean ± SD of four independent experiments. *P < 0.05 vs. control.
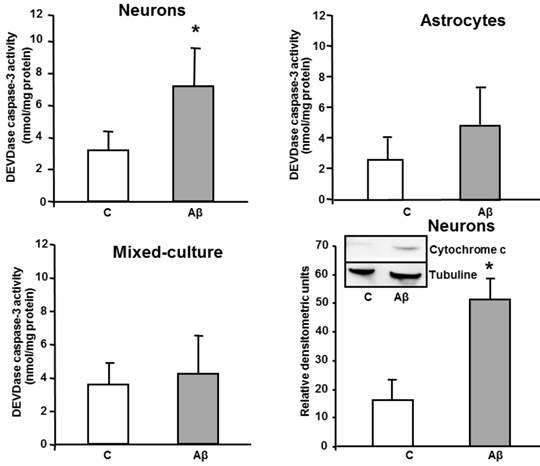
Aβ increases caspase 3 levels in neurons treated with Aβ toxic peptide
In order to investigate if these changes were part of an ongoing apoptotic process, we evaluated the effect of toxic Aβ1-42 treatment on activation of a late-effector caspase. In vitro activity of DEVDase caspase was assayed after treating cells with Aβ1-42 toxic peptide, and Aβ40-1 as a control peptide. As shown in Figure 2, the presence of the toxic peptide resulted in almost a 3 times increase in DEVDase caspase activity in primary cultured neurons, without significant changes in astrocytes or in mixed-culture cells, demonstrating apoptosis induction only in isolated neurons. One of the receptor-mediated mechanisms linked to activation of late caspases involves mitochondrial dysfunction, characterized by release of cytochrome c and subsequent activation of caspase 9 [17]. As shown in Figure 2, indeed Aβ1-42 causes cytochrome c release in cultured neurons.
In vitro DEVDase caspase-3 activity assay in astrocytes, neurons and mixed-culture cells. DEVDase caspase-3 activity was evaluated after 24 h incubation with 5 μM Aβ40-1 as a control (C) or Aβ1-42 as a toxic peptide (Aβ) in neurons, astrocytes and in mixed-culture cells. The results are representative of four independent experiments in quadruplicate. *P < 0.04 vs. control cells. Western-blot with Cytochrome c was evaluated in primary culture neurons. Data are mean ± SD of five independent experiments. *P < 0.05 vs. control.
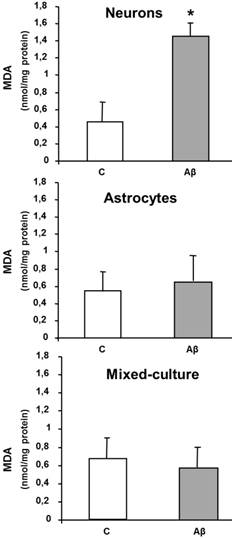
Aβ increases lipid peroxidation in neurons in primary culture
Aβ causes neurone cell death; likely by inducing caspases-dependent apoptosis (Figure 2), we also measured if this toxicity was associated with oxidative stress. For this purpose MDA levels were measured as a marker of lipid peroxidation. Aβ1-42 (5 µM x 24h) increased MDA levels in cultured neurons (Figure 3) as compared to controls. However the presence of Aβ1-42 did not modified either MDA levels in astrocytes or in mixed-culture cells (Figure 3). Only at higher concentrations of toxic peptide ≥ 20 µM we detected an increase of MDA levels in primary cultured astrocytes (data not shown).
MDA levels in neurons, astrocytes and mixed-culture cells. Cells were treated for 24 h with 5 μM Aβ40-1 (control, C) or 5 μM Aβ1-42 (Aβ). Data are mean ± SD of three different experiments. *P < 0.05 vs. control.
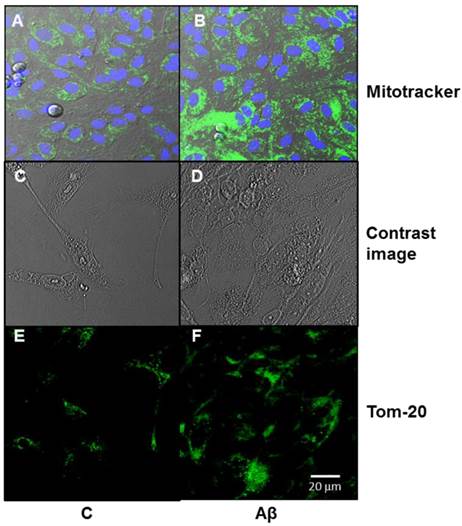
Aβ causes mitochondrial biogenesis in primary cultured of astrocytes
Figure 4 shows a significant increase in mitochondrial biogenesis in astrocytes incubated with the toxic peptide as compared to control. Panel A contains green images which are dispersed all over the astrocytes, whereas panel B displays brighter green spots which correspond to mitochondrial biogenesis (mitotracker). The blue dots in the images correspond to nuclei stained with Hoechst 33342. Panel E shows an immunofluorecent with Tom-20 antibody in control cells and in presence of Aβ1-42 peptide (panel F). The most immunofluorescent present in panel F indicates an increase in mitochondrial biogenesis in presence of Aβ1-42
Aβ1-42 increases mitochondrial aggregation in astrocytes in primary culture. Mitochondrial aggregation induced by Aβ1-42 was determined by confocal microscopy in astrocytes in primary culture treated for 6 h with 5 μM Aβ40-1 (C) or 5 μM Aβ1-42 (Aβ). Fluorescence products used were: Mitotracker (250 nM) to stain mitochondria and Hoechst 33342 (2 μg ml-1) to stain nuclei (A, B). Contrast image (C, D). Immunofluorescence was used with Tom 20 (E, F; 1:1000). Bar represents 20 μm.
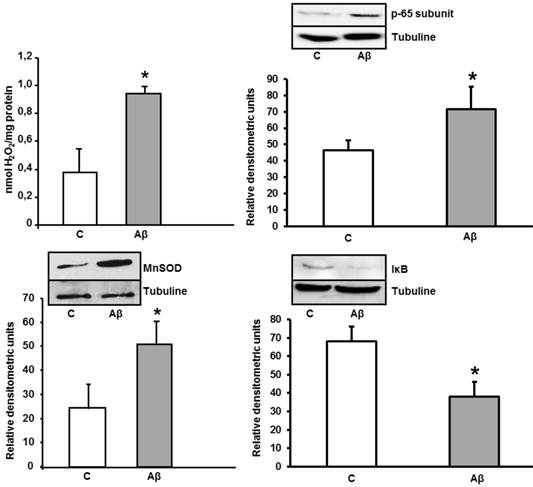
Aβ1-42 increases peroxide levels, MnSOD and p-65 (NF-κB) protein expressions in primary cultured astrocytes
To know whether astrocytes in primary culture present oxidative stress, we determined peroxide levels. After 24 h of incubation with Aβ1-42, we detected an increase of H2O2 levels in astrocytes in primary culture. Moreover, we also observed an important increase of MnSOD protein expression induced by ROS in cultured astrocytes (Figure 5), demonstrating an oxidative stress induction. We detect the nuclei-levels of p-65 protein, component of NF-κB in Figure 5 demonstrating an increase in p-65 protein expression in astrocytes after toxic peptide addition during 24 h. Furthermore, we noted a decrease in IκB protein level from cytoplasm astrocytes. Both results indicate that Aβ1-42 increases NF-κB protein expression in astrocytes.
Increase of peroxide levels, p-65 subunit (NF-κB), MnSOD, and IκB expressions caused by Aβ1-42 in astrocytes in primary culture. Peroxide levels were determined in cells treated for 24 h with 5 μM Aβ40-1 (C) or 5 μM Aβ1-42 (Aβ). Data are mean ± SD of three different experiments. *P < 0.01 vs. control. p-65, MnSOD and IκB protein expressions were determined in control conditions(C) and after addition of 5 μM toxic peptide Aβ1-42 during 24 h (Aβ). Data are mean ± SD of four independent experiments. *P < 0.05 vs. control.
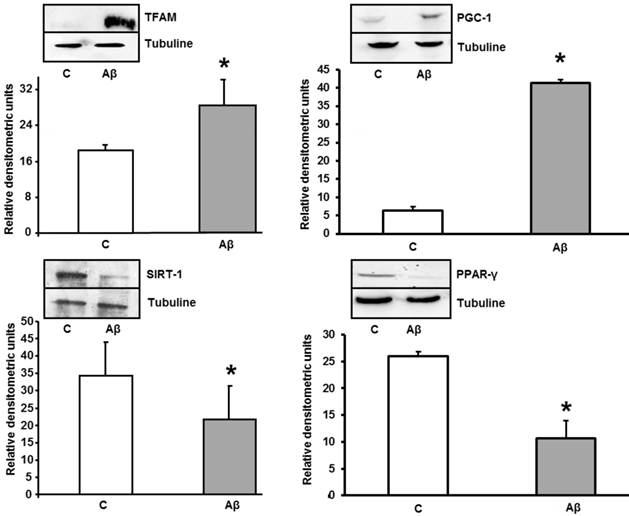
Aβ1-42 addition produced increase of TFAM and PGC-1 and decrease expression in PPAR-γ and SIRT-1 in primary cultured astrocytes
As mitochondrial biogenesis induction occurs, we determined the levels of TFAM and PGC-1 in the absence (control) and in the presence of Aβ1-42. Toxic peptide induces protein overexpression of TFAM and PGC-1 in astrocytes. TFAM and PGC-1 proteins are related with mitochondrial biogenesis in cells after mitochondrial stress. Figure 6 also shows a significant decrease in PPAR-γ (an anti-inflammatory protein) and SIRT-1 after treatment with Aβ1-42.
Aβ1-42 increases TFAM and PGC-1 and decreases PPAR-γ and SIRT-1 protein expressions in astrocytes in primary culture. Astrocytes were incubated with 5 μM of Aβ1-42 (Aβ) or 5 μM Aβ40-1 (C) for 24 h and collected to determine TFAM, PGC-1, PPAR-γ and Sirt-1 protein expressions by western blot. A representative immunoblot is shown in the top panel. Data are mean ± SD of four independent experiments. *P < 0.05 vs. control.
Discussion
The results of the present study show that Aβ1-42 peptide diminishes cellular viability, increases apoptosis and lipid peroxidation in neurons but neither in astrocytes nor mixed-culture. These findings indicate a protective role of astrocytes on neurons against Aβ1-42 toxic peptide. The results also demonstrate that astrocytes increase mitochondrial biogenesis and MnSOD protein expression; both mechanisms can contribute to the protective role of astrocytes. Relationship between neurons and astrocytes will be important to understand brain development and/or neurodegeneration. In brain from AD patients both inflammation and oxidative stress are increased [17], but their pathogenic significances remain unclear. Because anti-inflammatory therapy protects neurons against toxic substances [18], keep down inflammation and oxidative stress processes could improve neurodegeneration in AD. It is known that astrocytes have nutritional and support functions to neurons but also astrocytes can protect them against oxidative stress and inflammation [19, 20]. This protective effect may become deleterious when the activation of astrocytes increases [21]. Our results indicate that Aβ1-42 significantly increases MDA levels in neurons in primary culture without any changes in primary culture of astrocytes and mixed culture. Since MDA indicates lipid peroxidation, a reduction in MDA levels in mixed cultures is consistent with a protective effect of astrocytes against lipid peroxidation.
The presence of astrocytes in the mixed cultures might protect neurons to inflammation and oxidative stress caused by Aβ1-42, as authors published for other toxic components [22]. Carrero et al. [23] suggest that reactive astrocytes might be mediated by pro- and/or inflammatory gene expression and NFκB increase, which would potentiate neuronal demise. In this study, Aβ peptide induces damage by simple injection affecting neurons, astrocytes and blood vessels. The differences with Carrero et al. could be due to the different methodology used. Our experiments, carried out in cultured cells, allow us to study the effects of Aβ peptide on neurons, astrocytes or mixed-cultures selectively. Thus we could assess whether the neuronal damage is caused by direct effect of Aβ1-42 peptide or by inflammation generated in other cells. Moreover, increase of p-65 protein expression in astrocytes observed in our study could not indicate a neuronal demise but probably a protection of neurons. Although there is evidence that inflammation promotes AD, certain inflammatory mediators may also improve AD [20]. Wyss Corey et al. published the role of astrocytes such as defence cells inside the brain, and indicating a phagocyte process of astrocytes to eliminate and destroy Aβ peptide [20]. Our previous study also demonstrated the anti-inflammatory effects of astrocytes after Aβ-induction and protection with genistein or estradiol [6].
Furthermore, in cultured astrocytes we observed that Aβ1-42 upregulated MnSOD expression suggesting a possible antioxidant defence system that could protect neurons against oxidative damage scavenging ROS, such as occurs inside the brain [24]. Our results indicate that astrocytes protect neurons probably by an increase in mitochondrial biogenesis, obtaining better oxidative stress protection and perhaps producing control of inflammatory process. Increase reactive oxygen species is involved in mitochondrial DNA injury [9]. It is well known that TFAM induces mitochondrial DNA replication and maintenance [11]. In cultured SH-SY5Y cells, over-expression of TFAM diminished reactive oxygen species production and attenuated Aβ1-42-induced cell damage [25]. Accordingly, our results demonstrate an induction of p-65 (NF-κB) and over-expression of TFAM and PGC-1 after Aβ1-42 peptide addition in astrocytes in primary culture. Data indicate that mitochondrial biogenesis can be involved in neuronal protection against Aβ1-42 toxic peptide. These effects can be done by TFAM because over-expression of TFAM in astrocytes probably produces protection of mtDNA against Aβ1-42 peptide. TFAM can protect mtDNA from oxidative stress and inflammation by covering the mtDNA region [11, 13] as well as maintaining mtDNA copy number [26]. Furthermore, TFAM could increase mitochondrial biogenesis controlling mitochondrial dysfunction by initiate mtDNA transcription [27].
SIRT-1 has a role on deacetylate reactions of different proteins such as transcription factors involved in metabolic regulation, stress modulation, cell survival, aging and immune or inflammatory processes [28]. Expression of sirtuins is increased in caloric restriction of brain tissue [29] and a significant decrease in SIRT-1 protein has been noted in AD patients [30]. SIRT-1 modulates PGC-1 transcriptional expression and NF-κB activity in rat cardiac tissue [31]. Up or downregulation of PGC-1 determinates mitochondrial biogenesis through TFAM [32]. In our results, Aβ1-42 decreases SIRT-1 protein expression in cultured astrocytes which upregulates PGC-1 and TFAM resulting in augmentation of mitochondrial biogenesis. On the other hand we observed that Aβ1-42 diminishes PPAR-γ expression leading to an increase of NF-κB expression. This factor promotes PGC-1 phosphorylation that raises TFAM resulting in mitochondrial biogenesis (Figure 7). Taken together, our results indicate that Aβ1-42 exposition induces mitochondrial biogenesis in cultured astrocytes.
Signalling in astrocytes in primary culture after Aβ1-42 addition. The axis PGC-1-TFAM-Mitochondrial Biogenesis is regulated by PPAR-γ, NF-κB, SIRT-1 and MnSOD. Aβ1-42 toxic peptide decreases SIRT-1 and PPAR-γ and increases MnSOD and NF-κB leading to PGC-1-TFAM up-regulation resulting in mitochondrial biogenesis.
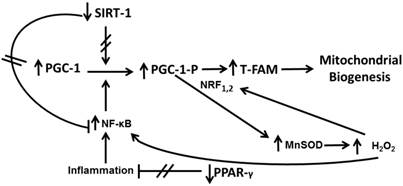
Astrocytes are involved in brain homeostasis, neural function, metabolic support to neurons and also in the synthesis of glutathione and its precursors [33]. The exact mechanisms as astrocytes protect neurons remain to be determined. Qin et al. have reported that mitochondrial biogenesis is altered in AD [34]. Because the presence of astrocytes prevents neuronal lipid peroxidation induced by Aβ1-42, we can hypothesize that increase of mitochondrial biogenesis in astrocytes supply high-energy demand to diminish free radical release induced by Aβ1-42 peptide. Therefore this mechanism could protect neurons against lipid peroxidation increase (Figure 7).
In conclusion, our results indicate that astrocytes may have a key role in protecting neurons against Aβ1-42 toxic peptide by increasing cell viability and mitochondrial biogenesis, obtaining better oxidative stress protection and perhaps modulating inflammatory processes. This might be a sign of a complex epigenetic process in Alzheimer's disease development.
Abbreviations
AD, Alzheimer's disease; Aβ, amyloid β; PPAR-γ, peroxisome proliferator activated receptor γ; TFAM, Mitochondrial transcription factor A; PGC-1, peroxisome proliferator-activated receptor γ coactivador 1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hardy JA, Higgins GA. Alzheimer´s disease: the amyloid cascade hypothesis. Science. 1992;256:184-185
2. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741-766
3. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787-795
4. Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145:1233-1248
5. Vallés SL, Borrás C, Furriol J. et al. Estradiol or genistein rescues neurons from Abeta-induced cell death by inhibiting activation of p38. Aging Cell. 2008;7:112-118
6. Vallés SL, Dolz-Gaiton P, Gambini J. et al. Oestradiol or genistein prevent Alzheimer´s disease-associated inflammation correlating with an increase PPARgamma expression in cultured astrocytes. Brain Research. 2010;1312:138-144
7. Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer´s disease. Trends Mol. Med. 2008;14:45-53
8. Sorensen L, Ekstrand M, Silva JP. et al. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci. 2001;21:8082-8090
9. Bozner P, Grishko V, LeDoux SP. et al. The amyloid beta protein induces oxidative damage of mitochondrial DNA. J Neuropathol Exp Neurol. 1997;56:1356-1362
10. Pappolla MA, Chyan YJ, Poeggeler B. et al. Alzheimer beta protein mediated oxidative damage of mitochondrial DNA: prevention by melatonin. J Pineal Res. 1999;27:226-229
11. Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7:39-44
12. Ikeuchi M, Matsusaka H, Kang D. et al. Overexpression of mitochondrial transcription factor A ameliorates mitochondrial deficencies and cardiac failure after myocardial infarction. Circulation. 2005;112:683-690
13. Hayashi Y, Yoshida M, Yamato M. et al. Reverse of age- dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci. 2008;28:8624-8634
14. Shangcheng X, Zhong M, Zhang L. et al. Overexpression of Tfam protects mitochondria against β-amyloid-induced oxidative damage in SH-SY5Y cells. FEBS Journal. 2009;276:3800-3809
15. Stokert JC, Blazquez-Castro A, Cañete M. et al. MTT assay for cell viability: Intracellular localization of the formazan product is in lipid droplets. Acta Histochem. 2012;114:785-796
16. Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg Biomembr. 1999;31:347-366
17. Cole GM, Morihara T, Lim GP. et al. NSAID and Antioxidant prevention of Alzheimer's Disease: Lessons from InVitro and Animal Models. Ann NY Acad Sci. 2004;1035:68-84
18. McGeer PL, Rogers J, McGeer EG. Inflammation anti-inflammatory agents and Alzeimer disease: the last 12 years. Alzheimer's Dis. 2006;9:271-276
19. Skaper SD. The brain as a target for inflammatory processes and neuroprotective strategies. Ann. N.Y. Acad. Sci. 2007;122:23-34
20. Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Medi. 2006;12:1005-1015
21. Griffin WS, Sheng JG, Royston MC. et al. Glial-neuronal interactions in Alzheimer's disease: the potential role of a 'cytokine cycle' in disease progression. Brain Pathol. 1998;8:65-72
22. Lööv C, Hillered L, Ebendal T. et al. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS One. 2012;7(3):e33090. doi: 10.1371/journal.pone.0033090
23. Carrero I, Gonzalo MR, Martin B. et al. Oligomers of β-amyloid protein (Aβ1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1β, tumour necrosis factor-α, and a nuclear factor κ-B mechanism in the rat brain. Exp. Neurol. 2012;236:215-227
24. Zhu X, Su B, Wang X. et al. Causes of oxidative stress in Alzheimer disease. Cell Mol. Life Sci. 2007;64:2002-2010
25. Xu S, Zhong M, Zhang L. et al. Overexpression of TFAM protects mitochondria against β-amyloid-induced oxidative damage in SH-SY5Y cells. FEBS Journal. 2009;276:3800-3809
26. Jeng JY, Yeh TS, Lee JW. et al. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J Cell Biochem. 2008;103:347-357
27. Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822-834
28. Chung S, Yao H, Caito S. et al. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch Biochem Biophys. 2010;501:79-90
29. Nisoli E, Tonello C, Cardile A. et al. Caloric restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314-317
30. Kumar R, Chaterjee P, Sharma PK. et al. Sirtuin1: a promising serum protein marker for early detection of Alzheimer's disease. PLoS One. 2013Apr16;8(4):e61560. Doi: 101371/journal.pone.0061560
31. Jian B, Yang S, Chen D. et al. Influence of aging and hemorrhage injury on Sirt1 expression: possible role of myc-Sirt1 regulation in mitochondrial function. Biochim Biophys Acta. 2011;1812:1446-51
32. Kang C, Ji LL. Role of PGC-1a signalling in skeletal muscle health and disease. Ann. N.Y. Acad. Sci. 2012;1271:110-117
33. Barreto GE, Gonzalez J, Torres Y. et al. Astrocytic-neuronal crosstalk: implications for neuroprotection from brain injury. Neurosci Res. 2011;71:107-113
34. Qin W, Haroutunian V, Katsel P. et al. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66:352-361
Author contact
![]() Corresponding author: Soraya L. Valles, Ph.D. Department of Physiology. School of Medicine. University of Valencia. Blasco Ibañez, 15. 46010 Valencia, Spain. Telf: 34-96-3864646. Fax: 34-96-3864642. Email: lilian.valleses.
Corresponding author: Soraya L. Valles, Ph.D. Department of Physiology. School of Medicine. University of Valencia. Blasco Ibañez, 15. 46010 Valencia, Spain. Telf: 34-96-3864646. Fax: 34-96-3864642. Email: lilian.valleses.