Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Acknowledgements
Conflicts of Interest
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(11):1089-1097. doi:10.7150/ijms.9239 This issue Cite
Research Paper
AKR1C3 Overexpression Mediates Methotrexate Resistance in Choriocarcinoma Cells
Jing Zhao1, Yang Xiang1 ![]() , Changji Xiao1, Peng Guo1, Dan Wang1, Ying Liu1, Yun Shen2
, Changji Xiao1, Peng Guo1, Dan Wang1, Ying Liu1, Yun Shen2
1. Departments of Obstetrics and Gynecology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People's Republic of China.
2. Reproductive Health Centre, National Science Institute for Family Planning, Beijing, People's Republic of China.
Received 2014-3-28; Accepted 2014-7-31; Published 2014-8-13
Abstract
Background: Chemotherapy is typically used to treat choriocarcinoma, but a small proportion of tumors develop resistance to chemotherapy. Similarly, methotrexate (MTX) is a first-line chemotherapy used to treat choriocarcinoma; although ~30% of patients are drug-resistant for MTX mono-therapy. Thus, we sought to elucidate the mechanism of chemotherapeutic-resistance of MTX.
Methods: RNA interference technology, colony formation, and MTT assays were used to investigate the role of aldo-keto reductase family 1, member C3 (AKR1C3) in MTX resistance in choriocarcinoma cells.
Results: AKR1C3 expression was higher in JeG-3R cells compared to JeG-3 cells and targeted inhibition of AKR1C3 expression with shRNA suppresses growth of choriocarcinoma cells as measured by colony formation and MTT assays. Overexpression of AKR1C3 increased chemotherapeutic resistance in JeG-3 cells. Furthermore, AKR1C3 silencing increases sensitivity to MTX in JeG-3R choriocarcinoma cells. Increasing MTX sensitivity spears to be related to DNA damage induction by increased reactive oxygen species (ROS), apoptosis, and cell cycle arrest.
Conclusions: Data show that AKR1C3 is critical to the development of methotrexate resistance in choriocarcinoma and suggest that AKR1C3 may potentially serve as a therapeutic marker for this disease.
Keywords: AKR1C3, Methotrexate (MTX), Choriocarcinoma cells, ROS, Chemotherapeutic-resistance.
Introduction
Choriocarcinoma is a malignant lesion often arising after molar pregnancies, but it can also occur after any form of gestation, including miscarriages and term pregnancies. The incidence of choriocarcinoma is varied worldwide, with significantly more occurrences in Asia and Latin America compared to Europe and North America [1]. The percentage of choriocarcinoma with metastases is high due to the propensity for vascular invasion. Recently, an 80-90% cure rate has made the disease one of the most dramatic successes in the treatment of human solid tumors by chemotherapy, largely due to methotrexate (MTX), actinomycin-D (KSM), and other chemotherapeutics [2-4]. Nevertheless, a few tumors maintain resistance to chemotherapy. Approximately 10-20% of low-risk patients without metastases and 30-50% of metastatic patients with low risk develop single-agent chemotherapeutic resistance and require multi-agent chemotherapy to achieve complete remission. Furthermore, ~20-30% of high-risk patients have an incomplete response to first-line multi-agent chemotherapy [5-8]. Therefore, chemotherapeutic resistance is the chief cause treatment failure. MTX is a first-line treatment of choriocarcinoma and is the single most commonly used agent in patients without metastases or with low risk of metastasis. In addition, MTX combined with other chemotherapy regimens is used in patients who are high risk, or drug resistance and recurrence. With serious acquired drug resistance, ~30% of patients develop drug-resistance for MTX mono-therapy [5], but how this occurs is uncertain.
Evidence suggests a potential role for estrogens in mediating placental trophoblastic growth and development [9,10] and choriocarcinoma cell proliferation and differentiation depends upon altered concentrations of E2 [10]. It is becoming increasingly clear that human aldo-keto reductases (AKRs) of the AKR1C family are intimately linked with cancer biology. In humans, 4 isoforms of AKR1C exist: AKR1C1, AKR1C2, AKR1C3, and AKR1C4. AKR1C3 functions as a synthase of 3α-HSD, 17β-HSD, 20α-HSD, and prostaglandin (PG) F, which catalyzes androgen, estrogen, progesterone, prostaglandin (PG), and xenobiotic metabolism [11-13]. Studies suggest that deregulated expression of AKR1C3 occurs in multiple types of cancers and contributes to the development of human cancer and chemotherapeutic resistance [14-19].
To identify a mechanism for chemotherapeutic resistance to MTX, we established a MTX-resistant JeG-3 choriocarcinoma cell sub line, JeG-3R, by sub-culturing JeG-3 cells with incremental and consecutive feeding [20]. We observed that expression of AKR1C3 is the highest among the AKR1C family in JeG-3R cells comparing with in JeG-3 cells. Furthermore, with RNA interference technology, AKR1C3 was contributed to MTX-induced drug resistance. Data indicate that suppression of reactive oxygen species (ROS) accumulation, which is enhanced in AKR1C3 overexpression cells, may underlie drug resistance to MTX in choriocarcinoma cells.
Materials and Methods
Cell culture
The chemo-resistant sub-line JeG-3R and the parental cell line JeG-3 [20] were grown in high glucose RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum (FBS, HyClone), 50 units/ml penicillin and 50 μg/ml streptomycin at 37°C in a humidified atmosphere of 95% O2 and 5% CO2.
Reverse transcription and quantitative polymerase chain reaction (RT-Q-PCR)
Total cellular RNA was extracted using an SV Total RNA Isolation kit (Promega Co., Madison, WI) according to the protocol provided by the manufacturer. One microgram of total RNA was reverse transcribed using a First Strand cDNA Synthesis Kit (TOYOBO, Japan) with random primers in a 20-μl reaction volume. Primers for PCR amplification were as follows: AKR1C1-forward: 5'-gtaagaaacggttgaactgg-3', AKR1C1-reverse: 5'-aaatcccaggacaggcatga-3'; AKR1C2-forward: 5'-tcacatgccattggttaacc-3', AKR1C2-reverse: 5'-acccggcttctattgccaat-3'; AKR1C3-forward: 5'-atttggcacctatgcacctc-3', AKR1C3-reverse: 5'-gaccagccttggaaaactca-3'; AKR1C4-forward: 5'-ctttcaaccacagatggtcc-3', AKR1C4-reverse: 5'-ctgcctgcagttgaagtttg-3'; GAPDH-forward: 5'-ccttggatgtggtagccgttt-3' and GAPDH-reverse: 5'-aactttcgatggtagtcgccg-3'. The Roche 480 sequence detection system was used with the following cycle parameters: Cycle 1 (95°C for 10 min); Cycle 2 (95°C for 15 s and 60°C for 1 min) × 40 cycles. Relative AKR1C3 mRNA were calculated as follows: ΔCT (sample) = CT (AKR1C3) - CT (GAPDH); Relative expression =2-ΔΔCT.
Western blot assays
Protein expression was compared by Western blot using mouse polyclonal anti-AKR1C3 (Sigma Co.). Briefly, cells were seeded in 6-well plates and cultured to 70% confluence. After the indicated treatments, cell protein extracts were prepared. Western blots were performed with 40 μg of protein extract and protein was detected using ECL chemiluminescent substrate (Amersham Pharmacia Biotech, Piscataway, NJ). GAPDH protein was used as a loading control. Western blot experiments were repeated at least three times.
Molecular cloning and transfection of shRNA in JeG-3R cells
Specific shRNA sequences for AKR1C3 were designed at the Sigma website. AKR1C3-targeting shRNA (AKR1C3-shRNA) and scrambled shRNA (scramble-shRNA) cassettes were constructed by PCR using the following primers: up-primer (hU6 promoter): TGGATCCaaggtcgggcaggaagag, down-primer-scramble: AGGATCCAAAAACAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGggtgtttcgtcctttccac; down-primer-AKR1C3: 5'-AGGATCCAAAAACCAGAGGTTCCGAGAAGTAAACTCGAGTTTACTTCTCGGAACCTCTGGCCGGTGTTTCGTCCTTTCCAC-3'. The resulting cassettes were then cloned into pGEM T easy vector using T4 DNA ligase (NEB, Beverly, MA) and confirmed by DNA sequencing. The TA cloning product was transiently transfected into JeG-3R cells using Lipofectamine 2000 reagent (Invitrogen) according to the protocol provided by the manufacturer.
Overexpression of AKR1C3
Human AKR1C3 cDNA was purchased (Origen, Cat. No: SC321532), and cloned into pcDNA3.1 and then transfected into JeG-3 cells using Lipofectamine 2000 reagent (Invitrogen).
Clonogenic assays
Monolayer cultures of control (scramble-JeG-3R) and AKR1C3 shRNA (AKR1C3-shRNA-JeG-3R) cells were treated with MTX (Sigma) at sequential concentrations (50 μM and then 100 μM) for 24 h; JeG-3 and AKR1C3 CDNA cell were treated with 25 μM and 50 μM MTX for 24 h. Then the cells were trypsinized and counted, and the appropriate number of cells were plated and incubated in six-well plates for 14-20 days. Colonies were stained with crystal violet (Sigma Chemical Co.), and colonies of 50 or more cells were counted.
Cell survival and proliferation assays
To evaluate the effect on proliferation after knock down of AKR1C3, cell growth inhibition was determined by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, 5 mg/ml) assay in 96-well plates. Transiently transfected AKR1C3-shRNA and scramble-shRNA cells were trypsinized and plated at a density of 3,000 cells/well in the 96-well plates. After incubation for 24 h, the cells were treated with or without 100 μM MTX. At various time intervals, absorbance was determined using a Multiskan plate reader at 490 nm.
Apoptosis/cell death detection assays
Monolayer cultures of exponential control (scramble- JeG-3R) and AKR1C3-shRNA- JeG-3R cells were treated with MTX (Sigma) at increasing concentrations (100 μM and then 200 μM) for 24 h. For the assessment of apoptosis and cell death, cells were analyzed using a FITC Annexin V Apoptosis Detection Kit 1 according to the protocol provided by the manufacturer (BD PharmingenTM). Briefly, treated cells were incubated with 0.5 mg/ml FITC-labeled recombinant Annexin V and 0.5 mg/ml propidium iodide (PI). Cells were then visualized and counted by flow cytometry. Cells that were FITC Annexin V positive and PI negative were considered to be in early apoptosis, and cells that were both FITC Annexin V and PI positive were considered to be in late apoptosis or already dead. Cell death was expressed as the percentage of Annexin-V and PI positive cells.
Cell cycle analysis by flow cytometry
Cells from scramble-JeG-3R and AKR1C3-shRNA groups were exposed to 100 μM MTX and then harvested at 0, 6, 12, and 24 h. Harvested cells were fixed in 75% ice-cold ethanol overnight, stained with propidium iodide, and counted with flow cytometry. Data were analyzed using ModFit software (Verity Software House, Topsham, MN).
ROS measurements
Flow cytometry measurements of 2', 7'-dichlorofluorescin diacetate (DCFH-DA) fluorescent probe were used to quantify cellular ROS. Cells from scramble-JeG-3R and AKR1C3-shRNA groups were exposed to 100 μM MTX for 24 h and 48 h. Monolayer cultures were incubated in 10 μM DCFH-DA for 30 min and then harvested. DCFH-DA-fluorescence was analyzed by flow cytometry (excitation 488 nm, and emission 525 nm) and con-focal microscopy. Mean fluorescent intensity was calculated after corrections for autofluorescence, and fold-changes were calculated relative to scramble-JeG3R cells.
Superoxide anion (O2-•) measurements
Cells from scramble-JeG-3R and AKR1C3-shRNA groups were exposed to 100 μM MTX for 24 h and 48 h and then measured the capacity of inhibition and produce O2-• by Inhibition and produce superoxide anion assay kit (Nanjing,China) according to the protocol provided by the manufacturer. Absorbance was read on a microplate reader at 550 nm. The production of superoxide anion unit =OD(treated OD-control OD)/(standard OD-control OD) ×0.15mg/ml×1000ml.
DNA damage assessment
DNA damage was assessed using the EpiQuikTM In Situ DNA Damage Assay Kit (Epigentek) according to the protocol provided by the manufacturer. Briefly, the phosphorylation of H2AX at serine 139, a sensitive marker of DNA damage, was detected by an anti-phosphor H2AX139 antibody. The ratio or amount of phosphor H2AX 139 was quantified with a horse radish peroxidase (HRP) conjugated secondary antibody-color development system and is proportional to the intensity of color development. Absorbance was read on a microplate reader at 450 nm. DNA damage % = OD (treated sample-blank)/OD (untreated sample-blank) ×100%.
Immunofluorescent assay
Cells from scramble-JeG-3R and AKR1C3-shRNA groups were plated onto 35 mm2 confocal dishes and treated with 100 μM MTX for 24 and 48 h. Cells were fixed in 4% paraformaldehyde (Sigma-Aldrich, St Louis, MO) for 10 min at room temperature and permeabilized in 0.1% Triton X-100 (Sigma-Aldrich) for 5 min. Thereafter, cells were blocked with 5% bovine serum albumin (BSA, Beyotime Biotechnology, Nanjing, China) for 3 h and then incubated with a primary antibody against γH2AX (Ser139, Cell Signaling Technology, MA) overnight at 4°C. Cells were rinsed three times with PBS for 5 min per wash before incubation with an Alexa Fluor 488-conjugated secondary antibody (Cell Signaling Technology) for 1 h. The cells were then exposed to 1 μg/ml 4',6-diamidino-2-phenylindole (DAPI, Beyotime Biotechnology) for nuclear DNA staining, rinsed three times with PBS (5 min per wash), and then visualized with confocal fluorescence microscopy (Leica Microsystems, Germany).
Statistical analysis
Numerical results are presented as the mean ± SD. A Student's t-test was performed to compare the means of different groups using SPSS 13.0 software (IBM SPSS Statistics). For all statistical analyses, a p value less than 0.05 was considered statistically significant.
Results
1. Up-regulated expression of AKR1C3 in MTX-resistant JeG-3R cells
To identify factors that contribute to the development of MTX resistance in choriocarcinoma, we performed serial culturing to establish an MTX-resistant derivative of JeG-3 cells, JeG-3R. RT-PCR results showed that the expression of AKR1C3 m RNA was the highest among the AKR1C family in JeG-3R cells comparing with JeG-3 cells (Fig. 1A).Given the predominant role of AKR1C3 in chemotherapeutic resistance [14-19], we further assessed the AKR1C3 expression with real-time PCR and Western blot. The results revealed up-regulated expression of AKR1C3 in JeG-3R cells. AKR1C3 mRNA was up-regulated 3.7-fold, and AKR1C3 protein was overexpressed compared with that in JeG-3 cells (Fig. 1B). These results suggest that AKR1C3 expression is enhanced by MTX.
AKR1C3 is up-regulated in MTX-resistant JeG-3R cells at the mRNA and protein level. AKR1C3 knockdown inhibits cell proliferation and survival, and enhances chemosensitivity in JeG-3R cells; overexpression of AKR1C3 increases chemo-resistance in JeG-3 cells.(A) RT-PCR showed that the expression of AKR1C3 mRNA was the highest among AKR1C family.(B) Real-time RT-PCR and western blot revealed that AKR1C3 is up-regulated in JeG-3R cells (*p<0.05). (C) Real-time RT-PCR and Western blot analysis were performed to compare AKR1C3 mRNA and protein levels in JeG-3R cells transfected with scramble-shRNA (control), or AKR1C3-shRNA. (D and E) Knockdown of AKR1C3 suppresses JeG-3R cell survival in a dose-dependent manner. The inhibitory effect of AKR1C3 knockdown on cell survival was determined by clonogenic assay and MTT assay (*p<0.05). (F) The cloning number was significantly higher in AKR1C3 CDNA cells than in the control cells.
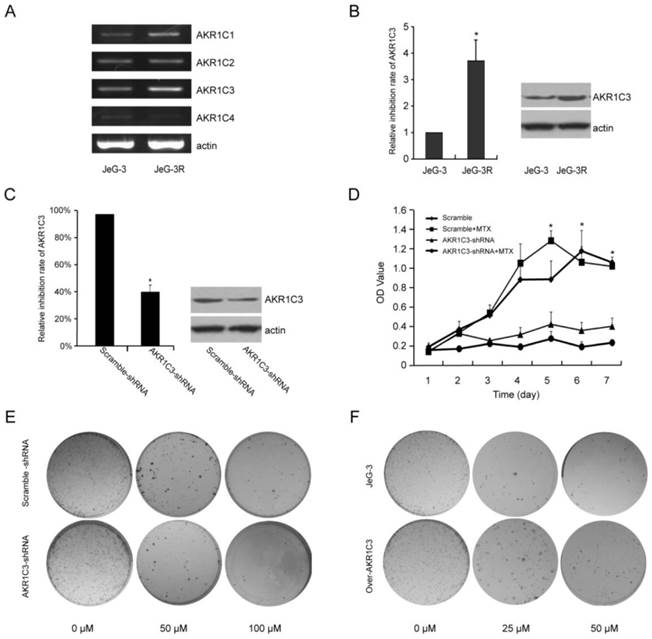
2. AKR1C3 silencing increases chemotherapeutic sensitivity to MTX in JeG-3R cells; overexpression of AKR1C3 increases chemo-resistance in JeG-3 cells
RNA interference technology was performed to assess the biological function of AKR1C3 in drug resistance induced by MTX. Compared with a scramble-shRNA, AKR1C3 expression in JeG-3R cells transfected with an AKR1C3-shRNA was decreased by 63% and 45% at mRNA and protein levels, respectively (Fig. 1C). Cell growth curves and cloning formation assays indicated that knockdown of AKR1C3 significantly increased the sensitivity of MTX to JeG-3R cells compared with controls (p<0.05, Fig. 1D, 1E). However, knockdown of AKR1C3 had no significant influence on cell proliferation (p>0.05). These results suggest that knockdown of AKR1C3 can reverse MTX drug resistance, which was not the result of cell proliferation inhibition. Overexpression of AKR1C3 increased chemo-resistance to MTX in JeG-3 cells (Fig. 1F).
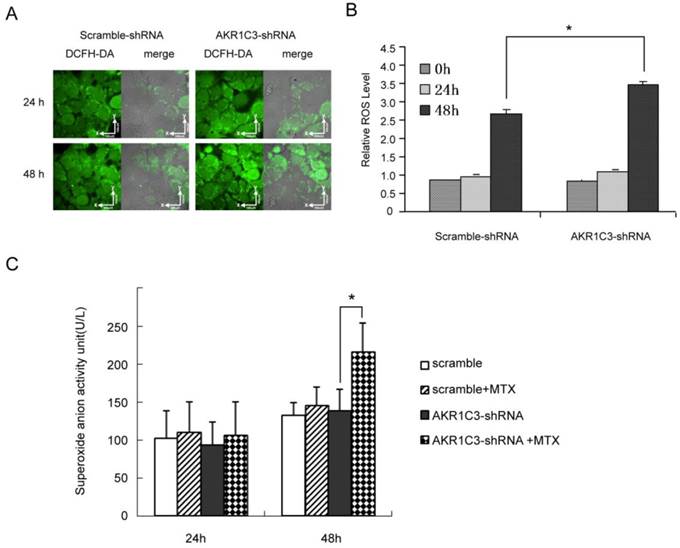
3. AKR1C3 silencing reverses MTX drug resistance by increasing ROS, especially increases superoxide anion
Cytotoxic drugs kill cancer cells by increasing ROS to cause apoptosis or necrosis. To further explore whether knockdown of AKR1C3 may reverse MTX resistance by regulating ROS-dependent cell death, we measured intracellular ROS using confocal microscopy and flow cytometry. The fluorescent intensity of 2', 7'-dichlorofluorescin (DCF), an indicator of ROS activity, was similar in JeG-3R cells transfected with AKR1C3-shRNA and scramble-shRNA controls. However, the fluorescent intensity increased gradually at 24 and 48 h after treatment with 100 µM MTX in AKR1C3-shRNA transfected cells and this increase in intensity was significantly higher than in scramble-shRNA transfected cells at 48 h (p<0.05, Fig. 2A, 2B).In addition, production of superoxide anion was significantly higher in AKR1C3-shRNA cells than that in scramble cells (p<0.05, Fig. 2C). These results suggest that knockdown of AKR1C3 increases MTX cytotoxicity by increasing intracellular ROS accumulation.
AKR1C3 knockdown can increase of ROS formation and production of much more superoxide anion. (A) JeG-3R cells transfected with scramble-shRNA or AKR1C3-shRNA were treated with 0 or 100 µM MTX for 24 or 48 h before ROS determination. The fluorescence intensity of intracellular DCF was detected by confocal microscopy as a measure of ROS activity. Results are representative of 3 independent experiments. (B) Flow cytometry was performed to determine the mean fluorescence intensity of intracellular DCF in scramble-shRNA and AKR1C3-shRNA transfected cells. Data are expressed as 1 of the control values and represent means±SD (n = 3; *p<0.05). (C) Production of superoxide anion was higher in AKR1C3-shRNA cells at 48h-MTX treatment than that in the control cells(*p<0.05),but there was no difference at 24h-MTX treatment.
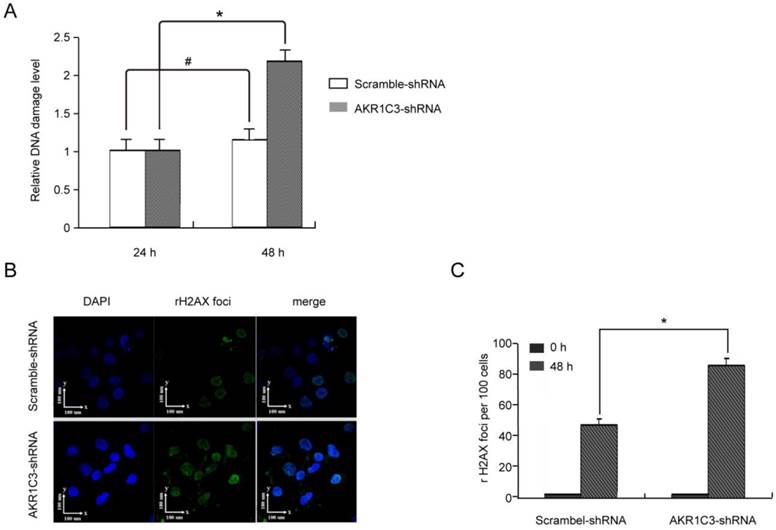
4. AKR1C3 silencing enhances cellular DNA damage by MTX and delays DNA repair
H2AXser139 phosphorylation is a sensitive marker of DNA damage. To determine whether knockdown of AKR1C3 affects the repair of DNA damage induced by MTX, we measured phosphorylated H2AXser139 in JeG-3R after transfection with AKR1C3-shRNA or scramble-shRNA. Assessment using the anti-phosphotyrosine antibody H2AXser139 (EpiQuikTM In Situ DNA Damage Assay Kit) with or without treatment with 100 µM MTX indicated that phosphorylated H2AXser139 was not significantly different between the two group at baseline and 24 h after treatment with MTX; however, at 48 h, phosphorylated H2AXser139 was significantly increased in AKR1C3-shRNA transfected cells compared with controls (p<0.05; Fig. 3A). These results were verified by immunofluorescent micrographs (Fig. 3B). The mean number of γH2AX foci per cell in the AKR1C3-shRNA group was significantly greater (p<0.05) than that in the control group (Fig. 3C). Our experiments suggest that expression of γH2AX foci is significantly increased in AKR1C3 knockdown cells 48 h after treatment with 100 µM MTX, indicating that AKR1C3 may play a protective role in MTX resistance in choriocarcinoma by preventing DNA damage.
AKR1C3 knockdown inhibition of DNA damage repair. The level of H2A.Xser139 phosphorylation was determined as a marker of DNA damage in JeG-3R cells transfected with scramble-shRNA and AKR1C3-shRNA following treatment with 0 or 100 µM MTX for 48 h. (A) Determination by EpiQuikTM In Situ DNA Damage Assay Kit. Data are expressed as 1 of control values and represent means±SD (n=3). (#, no difference; *p<0.05). (B) Determination by confocal microscopy. Representative immunofluorescence micrographs of γH2AX foci formation in AKR1C3-shRNA and scramble-shRNA JeG-3R cells are shown at 48 h after treatment with MTX. Results are representative of 3 independent experiments.(C) Counting the number of γH2AX foci observed in micrographs at the indicated time points demonstrated a significantly prolonged expression of γH2AX foci in AKR1C3-shRNA cells treatment with 100 µM MTX.
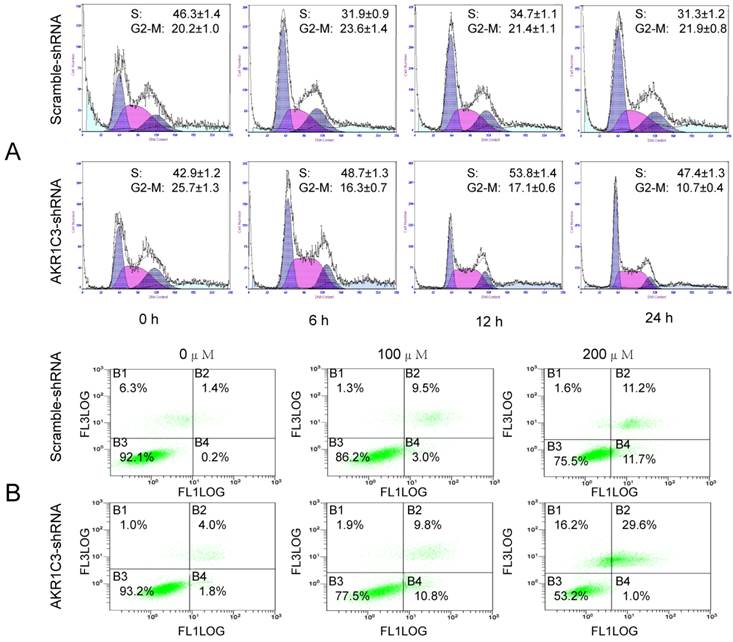
5. AKR1C3 silencing lead to cell cycle arrest and increased MTX-induced apoptosis
MTX is a cell cycle-specific agent mainly effecting the S phase. We observed effects of AKR1C3 knockdown on cell cycle distribution by flow cytometry. Significantly fewer G2/M phase cells were observed in AKR1C3-shRNA transfected cells compared to control scrambled shRNA cells at 6, 12, and 24 h after treatment with 100 μM MTX (Fig. 4A). Cell cycle arrest is known to induce apoptosis. Consequently, we performed Annexin V staining after treatment with different concentrations of MTX. Dead and apoptotic cells were significantly higher in the AKR1C3-shRNA group than that of the control group with increasing MTX concentration (p < 0.05, Fig. 4B).
AKR1C3 silencing induced of cell cycle arrest and apoptotic. (A) The cell cycle distributions of JeG-3R cells transfected with AKR1C3-shRNA and scramble-shRNA cells were determined following treatment with 100 µM MTX for 0, 6, 12, or 24 h. In AKR1C3-shRNA transfected cells, a sharp decrease in the fraction of cells in the G2/M phase was observed compared to the control. (B) JeG-3R cells transfected with scramble-shRNA or AKR1C3-shRNA were treated with 100 or 200 µM MTX for 24 h before assessment with a FITC Annexin V Apoptosis Detection Kit. Inhibition of AKR1C3 increased JeG-3R cell apoptosis in a dose-dependent manner. Treatment of AKR1C3-shRNA transfected cells with 100 µM MTX induced significantly more apoptosis compared to the control following treatment with 200 µM MTX.
Discussion
Despite new anticancer agents and more effective combination treatments, drug resistance remains a major obstacle to the successful treatment of advanced solid tumors. Therefore, new therapeutic targets are desperately needed. AKR1C3 is overexpressed in many human tumors and has been identified as a negative prognostic marker in various carcinomas, including advanced prostate cancer [15, 17, 21], breast cancer [22], and non-small cell lung cancer [23]. Further evidence indicating that AKR1C3 is an attractive target for cancer therapy is based on its association with tumor angiogenesis and resistance to cisplatin in colon cancer [19] and doxorubicin in breast cancer [18]. In the present study, we observed that AKR1C3 is overexpressed in MTX-resistant choriocarcinoma cell lines, and targeted inhibition of AKR1C3 expression using shRNA suppresses growth of choriocarcinoma cells. Our results demonstrate that AKR1C3 silencing increases MTX sensitivity in JeG-3R choriocarcinoma cells, suggesting that AKR1C3 may be a potential therapeutic marker in choriocarcinoma. The mechanism by which AKR1C3 shRNA increases MTX sensitivity may be related to elevated ROS and increased DNA damage, apoptosis, and cell cycle arrest induced upon silencing of AKR1C3.
AKR1C3 belongs to the aldo-keto reductase superfamily, which undergoes an oxidoreductase catalytic NADPH-dependent keto-aldehyde reaction. The metabolic byproduct of this reaction promotes DNA adducts or ROS formation, which leads to oxidative DNA damage. Inhibition of AKR1C3 expression has been reported to promote increased ROS and reversion of drug-resistance to cisplatin in colon cancer [19]. Consistently, in our work, we observed that intracellular ROS were significantly higher 48 h after treatment with MTX, and that this increase was enhanced in AKR1C3-silenced cells. ROS are generated as by-products of cellular metabolism, primarily in the mitochondria [24], and include free radicals such as superoxide anion (O2-•), hydrogen peroxide (H2O2), hydroxyl radical (HO·), nitric oxide (NO), and other species [25]. Recently, many cancer cell types have been shown to harbor increased ROS [26] which can elicit diverse responses depending on the magnitude, duration, and location of the exposure, as well as the specific ROS involved [27]. In general, low ROS is mitogenic and promotes cell proliferation and survival, whereas intermediate ROS causes transient or permanent cell cycle arrest and induces cell differentiation [28]. High concentrations of ROS can easily react with membrane lipids, altering membrane permeability; with DNA, causing damage and genomic instability; and with proteins, causing oxidative modifications that may produce less catalytically active enzymes or proteins that are more susceptible to proteolytic degradation [29, 30]. Normal breast epithelial stem cells have fewer ROS than more mature progenitor cells and therefore undergo less DNA injury during radiation, which allows them to preferentially escape radiation injury [31]. Increased production of ROS, especially increased superoxide anion in AKR1C3-shRNA cells, therefore, could explain the greater DNA damage and apoptosis after MTX exposure.
Cytotoxic drugs lead to cell death through a variety of mechanisms, including induction of apoptosis, necrosis, mitotic catastrophe, autophagy, and cell senescence. Apoptosis and mitotic catastrophe are chief mechanisms of chemotherapeutic cytotoxicity and MTX is a cell cycle-specific agent with a major role in the S phase, and can inhibit chromosomal replication and protein synthesis. Our data indicate that inhibition of AKR1C3 expression results in cell cycle arrest leading to apoptosis. Therefore, the reversal of MTX resistance by AKR1C3 may be mediated by cell cycle arrest caused by greater ROS. In addition, DNA strand breaks (due to increased ROS) can induce apoptotic signaling and cause irreversible oxidative DNA damage. It has been well-established that induction of DNA damage is a central mechanism through which many anticancer agents kill tumor cells [32-34]. With FITC Annexin V staining, we observed that more cells underwent apoptosis and dead cells increased significantly with increasing MTX concentrations in AKR1C3 knockdown cells. Furthermore, increased DNA damage was observed, as measured by the phosphorylation and formation of foci for γH2A.X, a rapid and sensitive marker for DNA damage induced by intracellular signal molecules. γH2A.X plays a critical role in maintaining genome integrity in eukaryotic cells, and its loss leads to genomic instability [35]. Of the various types of DNA damage, double-strand breaks are the most cytotoxic because of their potential to cause cell death and/or cell cycle arrest. Thus, an increased capacity of DNA damage repair in tumor cells is thought to be an important contributor to chemotherapeutic resistance during cancer treatments [36]. Phosphorylation of γH2A.X was recently reported to serve as an apoptotic induction signal [37]. These data suggest that DNA damage-induced apoptosis may contribute to the effects of AKR1C3 silencing in enhancing MTX sensitivity.
Conclusion
We report—for the first time—that AKR1C3 is activated in choriocarcinoma cells in response to MTX. Down-regulation of AKR1C3 promotes elevated ROS, which leads to DNA damage and facilitates apoptosis induced by cytotoxic chemotherapy. These results suggest that AKR1C3 may be a potential therapeutic target for chemotherapy sensitization in choriocarcinoma.
Acknowledgements
This work was supported by a grant from the National Nature and Science Foundation of China (Grant No. 81272890).
Conflicts of Interest
All authors have approved the manuscript and have no conflicts of interest to declare.
References
1. Song H, Wu B, Tang M. et al. Trophoblastic tumors: diagnosis and treatment. Beijing, China: Capital Hospital. 1981:57
2. Berkowitz RS, Goldstein DP. Current advances in the management of gestational trophoblastic disease. Gynecol Oncol. 2013Jan;128(1):3-5
3. Yang J, Xiang Y, Wan X, Yang X. Recurrent gestational trophoblastic tumor: management and risk factors for recurrence. Gynecol Oncol. 2006Nov;103(2):587-90
4. Powles T, Savage PM, Stebbing J. et al. A comparison of patients with relapsed and chemo-refractory gestational trophoblastic neoplasia. Br J Cancer. 2007Mar12;96(5):732-7
5. Abrão RA, de Andrade JM, Tiezzi DG, Marana HR, Candido dos Reis FJ, Clagnan WS. Treatment for low-risk gestational trophoblastic disease: comparison of single-agent methotrexate, dactinomycin and combination regimens. Gynecol Oncol. 2008Jan;108(1):149-53
6. Lurain JR, Nejad B. Secondary chemotherapy for high-risk gestational trophoblastic neoplasia. Gynecol Oncol. 2005May;97(2):618-23
7. Lurain JR, Singh DK, Schink JC. Primary treatment of metastatic high-risk gestational trophoblastic neoplasia with EMA-CO chemotherapy. J Reprod Med. 2006Oct;51(10):767-72
8. Ma Y, Xiang Y, Wan XR. et al. The prognostic analysis of 123 postpartum choriocarcinoma cases. Int J Gynecol Cancer. 2008;18(5):1097-101
9. Plessow D, Waldschläger J, Richter DU. et al. Effects of phytoestrogens on the trophoblast tumor cell lines BeWo and Jeg3. Anticancer Res. 2003;23(2A):1081-6
10. Jiang SW, Lloyd RV, Jin L, Eberhardt NL. Estrogen receptor expression and growth-promoting function in human choriocarcinoma cells. DNA Cell Biol. 1997Aug;16(8):969-77
11. Dufort I, Soucy P, Labrie F, Luu-The V. Molecular cloning of human type 3 3 alpha-hydroxysteroid dehydrogenase that differs from 20 alpha-hydroxysteroid dehydrogenase by seven amino acids. Biochem Biophys Res Commun. 1996Nov12;228(2):474-9
12. Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2,3a-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3a/17β-HSD activity and cellular distribution. Mol Endocrinol. 1997Dec;11(13):1971-84
13. Matsuura K, Shiraishi H, Hara A. et al. Identification of a principal mRNA species for human 3a-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998Nov;124(5):940-6
14. F. L. Khanim, R. E. Hayden, J. Birtwistle, et al. Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukemia. PLoS One. 2009Dec7;4(12):e8147
15. Fung KM, Samara ENS, Wong C. et al. Increased expression of type 2 3a-hydroxysteroid dehydrogenase/type 5 17b-hydroxysteroid dehydrogenase (AKR1C3) and its relationship with androgen receptor in prostate carcinoma. Endocr Relat Cancer. 2006Mar;13(1):169-80
16. Wako K, Kawasaki T, Yamana K. et al. Expression of androgen receptor through androgen-converting enzymes is associated with biological aggressiveness in prostate cancer. J Clin Pathol. 2008Apr;61(4):448-54
17. Stanbrough M, Bubley GJ, Ross K. et al. Increased expression of genes converting adrenal androgens to testosterone in androgenin dependent prostate cancer. Cancer Res. 2006;66(5):2815-25
18. Romana Novotna, Vladimir Wsol, Guangming Xiong, Edmund Maser. Inactivation of the anticancer drugs doxorubicin and oracin by aldo-keto reductase (AKR) 1C3. Toxicology Letters. 2008Sep;181(1):1-6
19. Toshiyuki Matsunaga, Aki Hojo, Yumi Yamane, Satoshi Endo, Ossama El-Kabbani, Akira Hara. Pathophysiological roles of aldo-keto reductases (AKR1C1 and AKR1C3) in development of cisplatin resistance in human colon cancers. Chem Biol Interact. 2013Feb25;202(1-3):234-42
20. Han B, Xiang Y, Wang Y, Wang Z, Zhang H, Huang S. Dihydrofolate reductase transcript level is not suitable for methotrexate-resistance prediction in choriocarcinoma cell line. Int J Gynecol Cancer. 2010Oct;20(7):1259-63
21. Nakamura Y, Suzuki T, Nakabayashi M. et al. In situ androgen producing enzymes in human prostate cancer. Endocr Relat Cancer. 2005Mar;12(1):101-7
22. Byrns MC, Duan L, Lee SH, Blair IA, Penning TM. Aldo-Keto Reductase 1C3 Expression in MCF-7 Cells Reveals Roles in Steroid Hormone and Prostaglandin Metabolism that may Explain its Over-Expression in Breast Cancer. J Steroid Biochem Mol Biol. 2010;118(3):177
23. Miller VL, Lin HK, Murugan P. et al. Aldo-keto reductase family 1 member C3 (AKR1C3) is expressed in adenocarcinoma and squamous cell carcinoma but not small cell carcinoma. Int J Clin Exp Pathol. 2012;5(4):278-289
24. Riess ML, Camara AK, Kevin LG, An J, Stowe DF. Reduced reactive O2 species formation and preserved mitochondrial NADH and [Ca2+] levels during short-term 17 degrees C ischemia in intact hearts. Cardiovascular Research. 2004;3(61):580-590
25. Lü JM, Lin PH, Yao Q, Chen C. Chemical and molecular mechanisms of antioxidants: experimental approaches and model systems. Journal of Cellular and Molecular Medicine. 2010;4(14):840-860
26. Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Lett. 1995Jan16;358(1):1-3
27. Storz P. Reactive oxygen species in tumor progression. Front Biosci. 2005May1;10:1881-96
28. Parola M, Bellomo G, Robino G, Barrera G, Dianzani MU. 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal. 1999;1(3):255-84
29. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000Nov9;408(6809):239-47
30. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002Jul;192(1):1-15
31. Diehn M, Cho RW, Lobo NA. et al. Association of Reactive Oxygen Species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780-783
32. te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002Mar15;62(6):1876-83
33. Jones KR, Elmore LW, Jackson-Cook C. et al. p53-dependent accelerated senescence induced by ionizing radiation in breast tumor cells. Int J Radiat Biol. 2005Jun;81(6):445-58
34. Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol. 2001;67:93-130
35. Bonner WM, Redon CE, Dickey JS. et al. GammaH2AX and cancer. Nat Rev Cancer. 2008Dec;8(12):957-67
36. Tobin LA, Robert C, Nagaria P. et al. Targeting abnormal DNA repair in therapy-resistant breast cancers. Mol Cancer Res. 2012Jan;10(1):96-107
37. Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009Apr2;458(7238):591-6
Author contact
![]() Corresponding author: Professor Yang Xiang, NO.1 Shuaifuyuan Wangfujing, DongCheng District, Beijing, China 100730. Tel: 86-10-6915-6287; Fax: 86-10-6512-4875 Email: xiangyang65com.
Corresponding author: Professor Yang Xiang, NO.1 Shuaifuyuan Wangfujing, DongCheng District, Beijing, China 100730. Tel: 86-10-6915-6287; Fax: 86-10-6512-4875 Email: xiangyang65com.