Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(10):988-993. doi:10.7150/ijms.8391 This issue Cite
Research Paper
Identification of de novo Mutations of Duchénnè/Becker Muscular Dystrophies in Southern Spain
Susana Garcia1, Tomás de Haro1,2, Mercedes Zafra-Ceres1, Antonio Poyatos1, Jose A. Gomez-Capilla1,2,3, Carolina Gomez-Llorente4 ![]()
1. UGC Laboratorios Clínicos. Hospital Universitario San Cecilio. Avd/Doctor Olóriz s/n 18012 Granada, Spain.
2. Instituto de Investigación Biosanitaria ibs. Granada, Spain.
3. Departamento de Bioquímica y Biología Molecular III e Inmunología. Facultad de Medicina. Universidad de Granada. Avd/ Madrid s/n 18071, Granada, Spain.
4. Departamento de Bioquímica y Biología Molecular II. Instituto de Nutrición y Tecnología de los Alimentos “José Mataix”. Centro de Investigaciones Biomédicas. Universidad de Granada. Avd/ Conocimiento s/n 18100 Armilla, Granada, Spain.
Received 2013-12-19; Accepted 2014-6-12; Published 2014-7-17
Abstract
Background: Duchénnè/Becker muscular dystrophies (DMD/BMD) are X-linked diseases, which are caused by a de novo gene mutation in one-third of affected males. The study objectives were to determine the incidence of DMD/BMD in Andalusia (Spain) and to establish the percentage of affected males in whom a de novo gene mutation was responsible.
Methods: Multiplex ligation-dependent probe amplification (MLPA) technology was applied to determine the incidence of DMD/BMD in 84 males with suspicion of the disease and 106 female relatives.
Results: Dystrophin gene exon deletion (89.5%) or duplication (10.5%) was detected in 38 of the 84 males by MLPA technology; de novo mutations account for 4 (16.7%) of the 24 mother-son pairs studied.
Conclusions: MLPA technology is adequate for the molecular diagnosis of DMD/BMD and establishes whether the mother carries the molecular alteration responsible for the disease, a highly relevant issue for genetic counseling.
Keywords: Duchénnè/Becker, Multiplex Ligation-dependent Probe Amplification (MLPA), de novo mutations.
Introduction
Duchénnè and Becker muscular dystrophies (DMD/BMD) are the most frequent genetic diseases among dystrophinopathies, which are caused by alterations of the dystrophin protein, essential in the interaction between cytoskeletal muscle fibers and extracellular matrix; however, although DMD and BMD are caused by mutations in the same gene, they have different ages of onset and distinct severity levels [1-3]. DMD is transmitted in a recessive X-linked fashion; the gene responsible (dystrophin gene [DMD]) is located in the short arm of chromosome X at locus 21 (Xp21), has a size of 2.4 Mb, and is formed by 79 exons. This disease affects males, while females are carriers, although some of them can manifest symptoms that vary from mild muscle weakness to a more severe clinical course [1]. Carrier females are at 50% risk of having an affected male or carrier female offspring.
The reported incidence is 1:3500 live newborn males for DMD and 1:18000 for BMD [1-3]. The disease is characterized by progressive symmetrical muscle weakness, which is earlier and more severe in DMD cases [2].
There are 4,000 known variations of the DMD gene (http://www.dmd.nl), of which the most important involve: the deletion of one or more exons (65% for Duchénnè and 85% for Becker); duplication of one or more exons (5-10% for DMD/BMD); or point mutations, small deletions, or insertions (25-30% for DMD and 5-10% for BMD) [4-7]. The two exon areas with the highest probability of deletions (hotspots) are between exons 44 and 53 and between exons 2 and 20 [4]. In 1935, Haldane reported that DMD/BMD is caused by sporadic mutations in one-third of the patients [8] and that this proportion depends on the patient's sex [9]. It is crucial to determine whether a de novo mutation or a mutation inherited from the mother is responsible for the disease, for which no effective treatment is available, in order to offer appropriate genetic counseling to the family of each affected male.
It has been known since 1987 that most of the mutations that cause DMD/BMD are preferentially found in 18 gene regions [10]; however, studies of these regions using different methods (southern-blot, quantitative PCR, multiplex amplifiable probe hybridization [MAPH], or fluorescent in situ hybridization [FISH]) proved able to detect alterations in only 20-30% of the 79 exons of the DMD gene [11-13]. The recently developed multiplex ligation-dependent probe amplification (MLPA) assay is a more useful technique for the study of index cases and possible carriers [14, 15], because it allows the detection of deletions and duplications of all exons of the gene [2, 13, 16-18]. However, it is important to confirm single exon deletions because of the possibility of small mutations (single nucleotide polymorphisms [SNPs], point mutations) preventing MLPA probe amplification [19].
The study objectives were to determine the incidence of DMD/BMD in Andalusia (Spain) and to establish the percentage of affected males in whom a de novo gene mutation was responsible, using the MLPA technique for molecular diagnosis of the disease in families with an affected male. In the present study population, de novo mutations were responsible for the disease in 16.7% of affected males. This is important information for providing correct genetic counseling.
Material and Methods
Patients
The study included all consecutive males referred to the Genetic Diagnosis Unit of the San Cecilio University Hospital (Granada) by hospitals in the Andalusia region between February 2005 and May 2013 for suspicion of DMD/BMD (ranging from isolated increases in creatine kinase [CK] and transaminases to a full set of DMD/BMD symptoms), along with one or more female relatives (up to 3rd degree relationship). The study was performed in accordance with the ethical principles of the Helsinki Declaration, and all participants or their guardians signed their informed consent for the genetic studies.
DNA isolation
DNA was isolated in a QIAcube extractor (Qiagen Iberia, S.L. Madrid, Spain) from peripheral blood samples drawn by venipuncture into tubes containing ethylenediaminetetraacetic acid (EDTA) as anticoagulant. DNA concentrations of samples were determined in a Jenway-Genove UV spectrophotometer (Jenway, Staffordshire, UK).
MLPA analysis
DMD gene mutations were studied in the extracted DNA samples by MLPA assay in accordance with the manufacturer's instructions (MRC Holland, Amsterdam, Holland). Briefly, DNA was denaturalized and hybridized for at least 12 h at 60 ºC with two probes: a probe containing oligonucleotides to analyze exons 1-10, 21-30, 41-50, and 61-70 (MIX 4); and a probe for exons 11-20, 31-40, 51-60, and 71-79 (MIX 5). Samples were treated with ligase for 15 min at 54 ºC, and the reaction was then stopped by incubation at 98 ºC for 5 min. Finally, the amplified product was mixed with a specific SALSA® product (oligonucleotide mixture) and underwent capillary electrophoresis in an ABI PRISM 310 genetic analyzer (Applied Biosystems, Foster City, CA). Results were compared to those obtained for three control samples in the same run. Gene Mapper software version 4.0 (Applied Biosystems, Foster City, CA) was used for the data analyses.
Calculation of the number of copies and preparation of Ratio Peak Area (RPA) graphs
Ratio peak areas (RPAs) were obtained for each run (MIX4 and MIX5) by dividing the area of the peak (A) corresponding to each exon by the sum of the areas (∑As) of the 45 peaks obtained in the sample electropherogram. The RPA for each exon (As/∑As) was then divided by the corresponding peaks obtained in the controls (Ac/∑As). All determinations for samples and controls were carried out in the same electrophoresis runs to yield comparable data. The RPA value is considered normal and reliable when it does not differ from the expected ratio by > 25%; the RPA is expected to be close to 1 in a healthy control. A sample is considered pathological in: a) males with one or more exons with RPA= 0 (deletion of corresponding exon), b) males with one or more exons with RPA =1.5-2.0 (duplication of corresponding exon), c) females with one or more exons with RPA ≈ 0.5 (deletion carrier), and d) females with one or more exons with RPA=1.5-1.75 (duplication carrier). The diagnosis is more reliable if the detected mutation affects two or more consecutive exons, because a single exon deletion can be caused by a polymorphism in the gene sequence that is complementary to the probe used to detect the deletion.
Results
The study included 190 participants, 84 males aged between 1 and 52 yrs (the 52-yr-old had BMD) and 106 females aged between 1 and 72 yrs. A total of 296 X chromosomes were analyzed. Out of the 84 males, DMD gene mutations were detected by MLPA assay in 38 (45.2%) but not in 46 (54.8%). Among the 46 males with no deletions or duplications: 5 were relatives of the index case with no myopathy symptoms; 15 had clinical (positive Gowers' sign, gait disorder), electromyographic, pathological, and/or biochemical data compatible with DMD/BMD, implying the possible presence of some type of mutation undetectable by MLPA, e.g., small deletions, insertions, or point mutations; 3 patients were finally diagnosed by molecular analysis with Steinert disease (CTG triplet determination by PCR amplification) and 1 with Landouzy Dejerine myopathy (Southern-blot of FSHD gene); and there were insufficient or missing clinical data in the remaining 22 patients (Table 1). Among the 38 males with MLPA-detected DMD gene mutation, 34 (89.5%) were diagnosed with exon deletions and 4 (10.5%) with exon duplications. Out of the 34 males with exon deletions, 4 showed single exon deletions. The diagnosis of these patients was confirmed by using a single PCR (data not shown), given the possibility of a false positive result due to changes in the sample DNA sequence detected by the probe (SNPs, point mutations).
Summary of patient data on family history, clinical symptoms, biochemical data, electromyogram, and muscle biopsy compatible with Duchénnè/Becker muscular dystrophy. Information on patients with other myopathies and with incomplete/missing clinical data.
| Age (yrs) | Family history | Clinical symptoms | Biochemical data | Electromyogram | Muscle biopsy | Clinical outcome | |
|---|---|---|---|---|---|---|---|
| 15 | - | Compatible with DMB | Elevated CK and transaminases | - | Partial absence of dystrophin | Currently in wheelchair; under mechanical ventilation program | |
| 1 | - | Compatible with DMD | Elevated CK | - | Absence of dystrophin | Motor skill regression; continuing in rehabilitation program | |
| 16 | - | Compatible with DMD | Elevated CK and transaminases | - | Absence of dystrophin | ||
| 10 | - | Compatible with DMD | Elevated CK and transaminases | Genetic analysis in external laboratory: presence of “codon stop” in DMD gene | |||
| 11 | Yes | Compatible with DMD | Elevated CK | - | Absence of dystrophin | - | |
| 10 | Yes | Compatible with DMD | Elevated CK | - | Absence of dystrophin | - | |
| 20 | - | Compatible with DMD | Elevated CK and transaminases | Myopathy signs | Absence of dystrophin | Currently in wheelchair | |
| 10 | - | Compatible with DMD | Elevated CK | - | - | Currently in wheelchair | |
| 44 | Yes | Compatible with DMB | Elevated CK | Myopathy signs | Partial absence of dystrophin | Currently in rehabilitation program | |
| 24 | Yes | Compatible with DMB | Elevated CK and transaminases | Myopathy signs | Partial absence of dystrophin | No clinical outcome data | |
| 22 | Yes | Compatible with DMB | Elevated CK and transaminases | Myopathy signs | Partial absence of dystrophin | No clinical outcome data | |
| 1.5 | - | Compatible with DMD | Elevated CK | Myopathy signs | Absence of dystrophin | Currently in rehabilitation program | |
| 21 | - | Compatible with DMD | Elevated CK and transaminases | - | Absence of dystrophin | Currently in wheelchair | |
| 9 | Yes | Compatible with DMB | Elevated CK | - | Partial absence of dystrophin | No clinical outcome data | |
| 3 | - | Compatible with DMD | Elevated CK and transaminases | - | Pending result of muscle biopsy | Currently in rehabilitation program | |
| Patients with diagnosis of other myopathies | Waist dystrophy (Landouzy Dejerine myopathy): 1 patient Myotonic dystrophy (Steinert's disease): 3 patients | ||||||
| Patients with incomplete/missing clinical data | 22 patients referred for non-specific symptoms, including: muscle weakness, cramps, hypertransaminasemia, elevated CK, unclear family history, and psycho-motor retardation. | ||||||
CK, creatine kinase; DMD, Duchénnè muscular dystrophy; DMB, Becker muscular dystrophy
Out of the 106 female relatives (to 3rd degree) studied, only 39 (36.8%) were mutation carriers; 35 of these (89.7%) carried exon deletions and 4 (10.3%) exon duplications. Table 2 displays the results of the 24 studies of mother-son pairs. The mutation detected in the son was absent in the mother in four of these pairs (16.7%), indicating the presence of a de novo mutation. The study of the mother was not possible in 14 of the 38 males diagnosed with gene deletion or duplication because the mother was dead (n=4) or not resident in Spain (n=2) or because the maternal sample was not delivered by the referring hospital.
Data on the 24 son-mother pairs studied.
| Affected exons in males | Affected exons in carrier mothers | |||
|---|---|---|---|---|
| Age (yrs) | Deletions | Duplications | Deletions | Duplications |
| 42 | 48-49 | - | 48-49 | - |
| 12 | 3-52-promotor | - | 3-52-promotor | - |
| 7 | 46-48 | - | 46-48 | - |
| 19 | 12-19 | - | 12-19 | - |
| 6 | 48-55 | - | 48-55 | - |
| 5 | 48 | - | 48 | - |
| 15 | 1-promotor | - | 1-promotor | - |
| 15 | 45 | - | 45 | - |
| 21 | 45-47 | - | 45-47 | - |
| 4 | 45-47 | - | Absent mutation | - |
| 19 | 3-7 | - | 3-7 | - |
| 3 | 45-52 | - | 45-52 | - |
| 1 | 3-7 | - | 3-7 | - |
| 10 | 3-4 | - | Absent mutation | - |
| 1 | 52 | - | 52 | - |
| 1 | 45-54 | - | 45-54 | - |
| 21 | 48 | - | 48 | - |
| 1 | 8-16 | - | Absent mutation | - |
| 15 | 61-77 | - | 61-77 | - |
| 6 | 49-50 | - | 49-50 | - |
| 5 | - | 2 | Absent mutation | - |
| 2 | 3-17 | - | 3-17 | - |
| 2 | 49-52 | - | 49-52 | - |
| 8 | 8-44 | - | 8-44 | - |
Figure 1 depicts the electropherograms and RPA graphs for a 7-yr-old male with DMD/BMD-compatible symptoms and family history and for his carrier mother.
Electropherograms of male DMD patient with deletion of exons 8 to 44 and of his mother, a carrier of the same deletions. The figure only shows the electropherogram for the run in which exons 1-10, 21-30, 41-50, and 61-70 are analyzed. Each peak represents an exon of the dystrophin gene. Arrows indicate the absence of signals corresponding to deleted exons 8, 9, 10, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 41, 42, 43, and 44. The ratio peak area (RPA) graphs alongside the electropherogram show an RPA for deleted exons of 0 for the son and 0.5 for the mother.
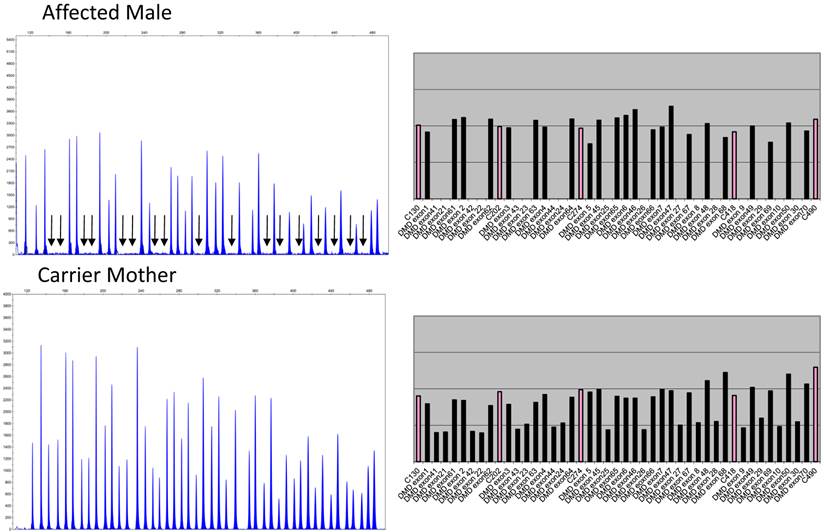
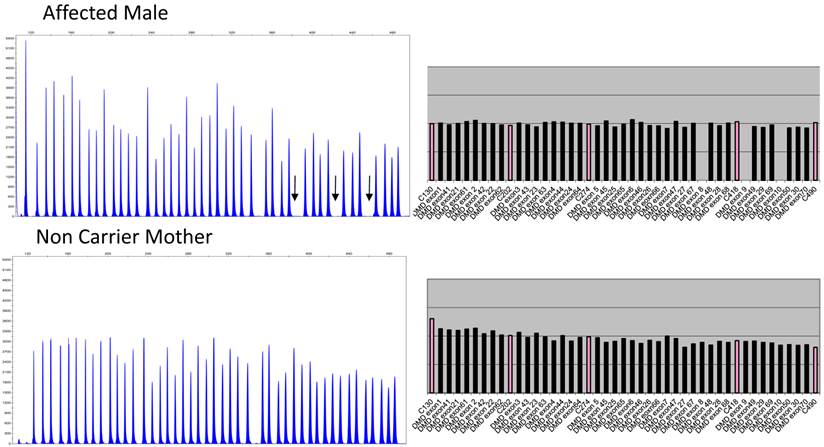
Figure 2 depicts the electropherograms and RPA graphs for a 3-yr-old male with DMD/BMD-compatible symptoms but no family history and for his non-carrier mother.
Electropherograms of male DMD patient with deletion of exons 8 to 10 and his non-deletion-carrying mother. The figure only shows the electropherogram for the run in which exons 1-10, 21-30, 41-50, and 61-70 are analyzed. Each peak represents an exon of the dystrophin gene. Arrows indicate the absence of signals corresponding to deleted exons 8, 9, and 10. The ratio peak area (RPA) graphs alongside the electropherograms show an RPA for deleted exons of 0 for the son and 1 for the mother.
Discussion
It has been known since 1987 that DMD/BMD is caused by mutations of the DMD gene, the largest known human gene (2.4 Mb and 79 exons) [20, 21]. The recently developed MLPA assay [14, 15] allows deletions and duplications to be detected in any of the 79 exons of this gene. We used this method to carry out a genetic study of DMD/BMD disease in 84 males. DMD gene mutations were detected in 38 of them and a further 15 were diagnosed with DMD/BMD based on clinical, electromyographic, and muscle biopsy data.
If we assume that the 15 males with DMD/BMD-compatible symptoms have mutations not detected by MLPA, it can be stated that the mutations in the 53 males with DMD/BMD were deletions (64.1%) or duplications (7.5%) or undetected small deletions, insertions, or point mutations (28.3%). These data are in agreement with previously published findings [5-7].
In 1935, Haldane claimed that a third of DMD/BMD cases involved spontaneous dystrophin gene mutations [8]. Findings since that date have been controversial, with a similar frequency being reported by some authors [21] but a higher [22] or lower frequency (around 20%) [23] by others. A sporadic mutation was detected in 16.7% of the present series of DMD/BMD patients. However, given the small sample size of 24 son-mother pairs, further investigation is warranted in a larger number of cases. Three out the four de novo mutations in the present study were deletions, one in the distal region and two in the proximal region, and there was only one duplication, consistent with suggestions that deletions are more frequent in de novo mutations than in other mutations [24].
There have been reports of both somatic [25] and germinal [26, 27] mosaicism in patients with dystrophinopathies. Knowledge of the carrier or non-carrier status of a woman is vital for genetic counseling because of the risk of recurrence from germinal mosaicism, recently estimated as 4.3% [28]. The lack of an effective treatment for this disease makes its prevention of paramount importance. In this context, prenatal diagnosis has offered women in DMD families the possibility of preventing the birth; however, as Helderman-van den Enden et al. recently pointed out in a study in the Netherlands, families with de novo mutations in the DMD gene cannot make use of prenatal diagnosis [29]. For this reason, it is essential not only to determine mutations in index cases but also to identify female carriers.
Furthermore, although most female carriers do not have symptoms of muscular disease, it is now known that a significant percentage suffer from muscle weakness and even dilated cardiomyopathy [30], and a five-yearly cardiac examination has been recommended by some authors [31]. There appears to be a wide consensus in favor of following up female carriers of DMD/BMD disease in order to rule out cardiac disease.
Although our results shed light on the proportion of de novo mutations in an Andalusian population, it is limited by the failure to include 14 of the 38 mother-son pairs traced and by some incomplete or missing clinical data for the others. However, given the very low incidence of this disease (1:3500 live newborn males for DMD and 1:18000 for BMD), the sample size achieved can be considered a study strength.
In conclusion, the MLPA assay is a sensitive, fast, and effective tool for diagnosing deletions and duplications of the dystrophin gene in affected males and carrier mothers. The assay offers a completely reliable diagnosis of the carrier or non-carrier status of the mother of a son with this disease, allowing appropriate genetic counseling to be offered with no need for muscle biopsy in most individuals with DMD/BMD.
Abbreviations
DMD/BMB: Duchénnè/Becker muscular dystrophy; MLPA: Multiplex Ligation dependent Probe Amplification.
Acknowledgements
The authors are grateful to all the males and their relatives that have participated in the present study and thank Richard Davies for assistance with English version. CGLL has a postdoctoral fellowship from the Plan Propio of the University of Granada.
Competing Interests
The authors declare no competing interest.
References
1. Moser H. Duchénnè muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet. 1984;66:17-40
2. Gatta V, Scarciolla O, Gaspari AR. et al. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum Genet. 2005Jun;117:92-98
3. Emery AE. Population frequencies of inherited neuromuscular diseases: a world survey. Neuromuscul Disord. 1991;1:19-29
4. Den Dunnen JT, Grootscholten PM, Bakker E. et al. Topography of the Duchénnè muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet. 1989;5:835-847
5. Hu XY, Ray PN, Murphy EG. et al. Duplicational mutation at the Duchénnè muscular dystrophy locus: its frequency, distribution, origin, and phenotype genotype correlation. Am J Hum Genet. 1990;46:682-695
6. Roberts RG, Bobrow M, Bentley DR. Point mutations in the dystrophin gene. Proc Natl Acad Sci USA. 1992;89:2331-2335
7. Aartsma-Rus A, Van Deutekom JC, Fokkema IF. et al. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135-144
8. Haldane JBS. The rate of spontaneous mutation of a human gene. J Genet. 1935;31:317-326
9. Haldane JBS. Mutation in the X-linked recessive type of muscular dystrophy: a possible sex difference. Ann Hum Genet. 1956;20:344-347
10. Koenig M, Hoffman EP, Bertelson CJ. et al. Complete cloning of the Duchénnè muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509-517
11. Chamberlain JS, Gibbs RA, Ranier JE. et al. Deletion screening of the Duchénnè muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988;16:11141-11156
12. Beggs AH, Koenig M, Boyce FM. et al. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet. 1990;86:45-48
13. White S, Kalf M, Liu Q. et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet. 2002;71:365-374
14. Schouten JP, McElgunn CJ, Waaijer R. et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57
15. Sellner LN, Taylor GR. MLPA and MAPH: new techniques for detection of gene deletions. Hum Mutat. 2004;23:413-419
16. Janssen B, Hartmann C, Scholz V. et al. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: potential and pitfalls. Neurogenetics. 2005;6:29-35
17. Ligon AH, Kashork C, Richards CS. et al. Identification of female carriers for Duchenne and Becker muscular dystrophies using a FISH-based approach. Eur J Hum Genet. 2000;8:293-8
18. Schwartz M, Duno M. Improved molecular diagnosis of dystrophin gene mutations using multiplex ligation-dependent probe amplification method. Genet Test. 2004;8:361-367
19. Hills A, Ahn JW, Donaghue C. et al. MLPA for confirmation of array CGH results and determination of inheritance. Mol Cytogenet. 2010;3:19-26
20. Hoffmann EP; Brown RH, Kunkel LM. Dystrophin: The protein product of the Duchénnè muscular dystrophy locus. Cell. 1987;51:919-928
21. Caskey CT, Nussbaum R, Cohan L. et al. Sporadic occurrence of Duchénnè muscular dystrophy: evidence for new mutation. Clin Genet. 1980;18:329-341
22. Lane R, Robinow M, Roses A. The genetic status of mothers of isolated cases of Duchénnè muscular dystrophy. J Med Genet. 1983;20:1-11
23. Barbujani G, Russo A, Danieli G. et al. Segregation analysis of 1885 DMD families: significant departure from the expected proportion of sporadic cases. Hum Genet. 1990;84:522-526
24. Lee T, Takeshima Y, Kusunoki N. et al. Differences in carrier frequency between mothers of Duchenne and Becker muscular dystrophy patients. J Human Genet. 2014;59:46-50
25. Kesari A, Neel R, Wagoner L. et al. Somatic mosaicism for Duchénnè dystrophy: evidence for genetic normalization mitigating muscle symptoms. Am J Med Genet A. 2009;149A:1499-1503
26. Bakker E, Vanbroeckhoven C, Bonten EJ. et al. Germline Mosaicism and Duchénnè Muscular-Dystrophy Mutations. Nature. 1987;329:554-556
27. Bakker E, Veenema H, den Dunnen JT. et al. Germinal mosaicism increases the recurrence risk for 'new' Duchénnè muscular dystrophy mutations. J Med Genet. 1989;26:553-559
28. Helderman-van den Enden AT, de Jong R, den Dunnen JT. et al. Recurrence risk due to germ line mosaicism: Duchénnè and Becker muscular dystrophy. Clin Genet. 2009;75:465-472
29. Helderman-van den Enden AT, Madan K, Breuning MH. et al. An urgent need for a change in policy revealed by a study on prenatal testing for Duchenne muscular dystrophy. E J Human Gene.t. 2013;21:21-26
30. Hoogerwaard EM, van der Wouw PA, Wilde AA. et al. Cardiac involvement in carriers of Duchénnè and Becker muscular dystrophy. Neuromuscul Disord. 1990;9:347-351
31. Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy; 7th-9th June 2002, Naarden, the Netherlands. Neuromuscul Disord 13:166-172.
Author contact
![]() Corresponding author: Dr. Carolina Gomez-Llorente, Department of Biochemistry and Molecular Biology II, Institute of Nutrition and Food Technology. Center of Biomedical Research, University of Granada. Avd. Conocimiento s/n 18100, Armilla, Granada, Spain. Tel +34 958241000 ext. 20322 Fax: +34958243391 E-mail: gomezlles.
Corresponding author: Dr. Carolina Gomez-Llorente, Department of Biochemistry and Molecular Biology II, Institute of Nutrition and Food Technology. Center of Biomedical Research, University of Granada. Avd. Conocimiento s/n 18100, Armilla, Granada, Spain. Tel +34 958241000 ext. 20322 Fax: +34958243391 E-mail: gomezlles.