Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(9):870-879. doi:10.7150/ijms.8758 This issue Cite
Research Paper
Suppression of Peritoneal Tumorigenesis by Placenta-Derived Mesenchymal Stem Cells Expressing Endostatin on Colorectal Cancer
Dongmei Zhang1†, Lan Zheng2†, Huashan Shi1, Xiancheng Chen1, Yang Wan1, Hailong Zhang1, Meng Li1, Lian Lu1, Shuntao Luo1, Tao Yin1, Honggang Lin1, Shasha He1, Yan Luo1 ![]() , Li Yang1
, Li Yang1 ![]()
1. State Key Laboratory of Biotherapy/Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu, 610041, P. R. China
2. Department of Obstetrics and Gynecology, Second West China Hospital of Sichuan University, Chengdu, 610041, P. R. China
† Equal contributors.
Received 2014-2-4; Accepted 2014-5-11; Published 2014-6-13
Abstract
MSCs-based therapy for cancer is a relatively new but rapidly growing area of research. Human term placenta, an attractive source of MSCs (PMSCs), appears to have great advantage due to its easy access without invasive procedures, its lack of ethical issues and its high-throughput and young age. In the present study, we isolated MSCs from placenta and characterized their morphology and differentiation capacities. We next investigated the underlying antitumor effects and the potential mechanism of PMSCs to express endostatin using adenoviral transduction (Ad-Endo) in a colorectal peritoneal carcinomatosis (CRPC) mouse model. For in vitro experiments, the migratory potential of Ad-Endo-PMSCs towards tumor cells was demonstrated using a double-chamber assay, and the anti-angiogenesis ability of endostatin from engineered PMSCs was evaluated using the tube formation assay. For the in vivo study, mice harboring CT26 colorectal cancer indicated a significant reduction in tumor nodules and a prolongation of survival following Ad-Endo-PMSCs therapy. These observations were associated with significantly decreased tumor cell proliferation and blood vessel counts as well as increased tumor cell apoptosis. These data suggested the potential of PMSCs-based gene therapy for the targeted delivery of therapeutic proteins in cancer.
Keywords: placenta-derived mesenchymal stem cells, adenovirus vector, gene therapies, endostatin, colorectal cancer
Introduction
Colorectal peritoneal carcinomatosis (CRPC) is one of the most commonly diagnosed cancers in the world and the third leading cause of death worldwide [1]. According to data published by the International Research on Cancer (IARC), approximately 1 million new cases of CRPC are diagnosed each year, and 500,000 deaths are due to this disease. Despite improved treatment models, CRPC remains unresponsive to traditional therapy. Approximately 8% of patients have been diagnosed with surgically induced metastasis, and 25% of patients experience recurrence of the primary CRPC [2, 3]. Thus, gene therapy represents a novel and promising approach for cancer treatment.
Invasion and metastasis are the major causes of CRPC treatment failure. The current wisdom is that tumor growth, invasion and metastasis are angiogenesis dependent [4, 5]. In the early 1970s, Judah Folkman, a highly respected pioneer and researcher in this field, hypothesized that new blood vessels were essential for tumor growth and that an anti-angiogenic strategy might constitute a new therapeutic approach for solid tumors [6]. Over the past several decades, the central role of angiogenesis in the process of tumor growth and dissemination has been well established [7]. Currently, over 60 angiogenesis inhibitors have been identified [8]. Among these molecules, endostatin is the first endogenous angiogenesis inhibitor to be introduced into clinical trials. Several studies have indicated that endostatin exhibits a broad spectrum of antitumor activity with low toxicity [9-11]. However, due to the rapid turnover of endostatin in the blood, long-term administration of the inhibitor is required to ensure the suppression of tumor growth in vivo [12]. Moreover, it has proven to be extremely difficult to produce the functional polypeptide; thus, administration of endostatin remains expensive and places a burden on the patients [12].
MSCs are considered one of the most exciting platforms for cell-based gene therapy, and currently, more than 150 clinical trials around the world use MSCs for tissue repair, hematopoietic stem cell transplantation and autoimmune diseases. More recently, the ability of MSCs to target to tumor locations has been demonstrated in a relatively large number of pre-clinical studies, revealing the potential of MSCs for use as an attractive vehicle for the delivery of anticancer agents [13, 14]. Previous studies have demonstrated the strong antitumor effect of MSCs as gene delivery vectors. Komarova S et al. reported that MSCs with forced expression of oncolytic adenoviruses exhibited strong inhibitory growth on ovarian cancer in vivo [15]. Nakamura K et al. observed that intratumoral injection of MSCs modified with IL-2 targeted to sites of tumorigenesis and potently inhibited the growth of malignant gliomas[16]. MSCs can be isolated from several adult and fetal tissues, such as bone marrow, adipose tissue, heart, skeletal muscle, pancreas, placenta, cord blood and circulating blood. Although bone marrow is the initial and traditional source of MSCs (BMSCs), the collection of MSCs is invasive, with only 1-20/105 MSCs obtained from bone marrow [17, 18]. In addition, the number and differentiation capacity of BMSCs decreases dramatically with age [19]. The placenta, as a materno-fetal organ, originates within the first stages of embryological development and has drawn great interest as a source of MSCs for clinical research. Previous studies have shown that no significant difference could be detected between placenta-derived mesenchymal stem cells (PMSCs) and BMSCs in terms of morphology, cell-surface antigen expression, chemokine receptor display, differentiation capacity and immunosuppressive ability [20, 21]. In addition, as a dispensable tissue after the delivery of a baby, the placenta can provide abundant MSCs without invasive procedures or ethical issues [22]. Thus, the placenta may represent a pool of MSCs and may serve as a preferred source of MSCs for future clinical use.
In the present study, we investigated the anti-tumor activities of Ad-Endo-transfected PMSCs via peritoneal administration in a CT26 CRPC model. Our data indicated that PMSCs transfected with Ad-Endo could effectively suppress tumor growth and significantly improve host survival. Thus, PMSCs may be a safe and effective gene delivery system for further clinical application.
Materials and Methods
Mice
Female Balb/c mice 6-10 weeks of age were purchased from Peking Union Medical College. The animals were maintained in polycarbonate cages, with a dedicated aseptic environment. During the experimental period, all of the research protocols were approved by the Institute's Animal Care and Use Committee of Sichuan University.
Cell lines and culture
CT26 colorectal cancer cell line and transformed human embryonic kidney cell line 293T cells were purchased from ATCC (American Type Culture Collection, Rockville, MD). CT26 were cultured in RPMI1640 (Gibco BRL, Grand Island, N.Y.). 293T were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL, Grand Island, N.Y.), PMSCs were cultured in DMEM-low glucose (DMEM-LG) (Gibco BRL, Grand Island, N.Y.). A detailed protocol regarding the isolation of PMSCs has been previously published in PLOS ONE[23]. The medium was supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco, Melbourne, Australia) and 1% penicillin/streptomycin (Gibco, Melbourne, Australia). All cell lines were maintained in 95% air humidified atmosphere with 5% CO2 at 37℃.
Cell surface phenotype
To determine the surface antigens of PMSCs, cells at Passage 5 (P5) were collected and stained using various mouse anti-human monoclonal antibodies conjugated with fluorescein isothiocyanate (FITC): CD166, CD29, CD90, CD105, and CD34 (Invitrogen, Thermo, Millipore, Neomarkers, eBioscience). The same FITC-labeled mouse IgG (BD Biosciences, USA) without primary antibodies served as a negative control. At least 104 events were analyzed using flow cytometry.
Multipotent differentiation of PMSCs
To assess the multipotent differentiation capacity of PMSCs, experiments including osteogenic, adipogenic and chondrogenic induction were performed in vitro.
Osteogenic differentiation was induced by culturing cells in DMEM containing 10% FBS, 0.1 μM dexamethasone, 10 mM β-glycerophosphate and 50 μg/ml ascorbate-2 (Sigma-Aldrich, USA). The medium was changed twice a week, and 2 weeks later, calcium depositions were evaluated using Alizarin red S staining (Sigma-Aldrich, USA) [24].
For adipogenic differentiation, 1×105 PMSCs were seeded onto a 6-well plate. When cells were 70-80% confluent, the medium was replaced with DMEM containing 10% FBS, 0.5 μM isobutyl-methylxanthine, 0.25 μM dexamethasone, 10 μM insulin and 50 μM indomethacin (Sigma-Aldrich, USA). The induction medium was refreshed twice a week. Fourteen days later, lipid vesicles were observed using Oil-Red-O staining [25].
For chondrogenic differentiation, transwell dishes with 6.5-mm diameters and 0.4-μm pores were used (Millipore, Billerica, MA, USA). Next, 1×105 PMSCs were seeded on the top chamber and centrifuged in a 24-well plate (200 g). Five minutes later, 500 μl of StemXVivo chondrogenic differentiation medium (CCM005, CCM006, R&D, USA) was added to the lower well of the 24-well plate. The medium was changed twice a week. Twenty-one days later, chondrogenic differentiation was assessed using Alcian blue staining (Sigma-Aldrich, USA) [26].
PMSCs transfection
Low expression of the Coxsackie-adenovirus receptor (CAR) is a limiting factor for the transduction efficiency of PMSCs with Ad vectors. In this study, we used Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for Ad-Endo infection. Briefly, 1×105 PMSCs were seeded onto a 6-well plate, and the next day, Ad-EGFP was transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) at a serial multiplicity of infection (MOI=1500, 3000). The virus/Lipofectamine 2000 ratio was 2.5×108 vp/µg. Forty-eight hours later, the transduction efficiency was analyzed using an inverted microscope and flow cytometer, and transfection without Lipofectamine 2000 was used as a control.
Western Blotting Analysis
Western blotting analysis was performed as previously described (17). Briefly, 1×105 PMSCs were transfected with Ad-Endo at an MOI of 3000 (Ad-Endo=3×108 vp, Lipofectamine 2000=1.2 µg). After 48 h, the conditioned media (CM) was harvested and concentrated by ultrafiltration (Centricon YM-3; Millipore, Bedford, MA, USA). Proteins were separated using 12% SDS-PAGE gel and transferred onto PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked in 5% non-fat milk (Biorad, USA) and then incubated with an anti-endostatin antibody (Santa Cruz Biotechnology, CA) (1:500) followed by horseradish peroxidase-conjugated anti-rabbit immunoglobulins (Sigma-Aldrich, USA) (1:5000). β-actin (1:2000) was used as a loading control, and bands were analyzed using an enhanced chemiluminescence detection system (Thermo Fisher Scientific Inc. USA).
In vitro migration assay
To analyze the ability of PMSCs to target tumors, transwell dishes with 8-μm pore filters were used (PI8P01250, Millipore, Billerica, MA, USA). Then, 1×104 PMSCs or Ad-Endo-PMSCs were seeded on the top chamber of the transwell filter, with 2×104 CT26 cells or 293T cells on the 24-well plate. After incubation for 24 h, the cells were fixed with methanol for 15 min and stained using Crystal Violet (Sigma-Aldrich, USA). Finally, the membrane was transferred onto a cover slide, and the number of migrated cells from five random fields was quantified using light microscopy [27]. All experiments were performed in triplicate.
In vitro tube formation assay
The tube formation assay was performed as previously described [28]. Briefly, 2×104 human umbilical vein endothelial cells (HUVECs) were suspended in 200 μl of CM and the supernatant obtained from Ad-Null-PMSCs and untransfected PMSCs. Fresh DMEM-LG was used as a negative control. Next, the cells were seeded onto the matrigel (BD Biosciences, USA) and maintained in 95% air humidified atmosphere with 5% CO2 at 37℃. After 6 h, the cells were imaged using a digital camera attached to an inverted microscope.
CT26 colorectal peritoneal carcinomatosis model
Six-week-old female Balb/c mice were randomly divided into four groups (10 mice per group). Next, 3.0×105 CT26 cells were injected into the abdominal cavity of the mice. After 4 days, these mice were intraperitoneally administered with 0.9% NaCl (Control), PMSCs (1.0×105 cells), Ad-Null-PMSCs (1.0×105 cells transfected with 3.0×108 vp Ad-Null) or Ad-Endo-PMSCs (1.0×105 cells transfected with 3.0×108 vp Ad-Endo) individually every 4 days for a total of four administrations. The mice in all of the groups were sacrificed when the mice in the Control group became moribund. The size and number of tumor nodes were determined. In addition, another subgroup of mice within the same group division was monitored daily for 60 days.
Immunohistochemistry and TUNEL assay
For immunofluorescent staining, frozen tissue was sectioned (8 µm) and stained with CD31 (1:100) or Ki67 (1:100) (ab28364, ab15580, Abcam, UK), and the nuclear counter stain 4',6-diamidino-2-phenylindole (DAPI). A TUNEL assay (Promega, USA) was performed according to the manufacturer's instructions. Images were captured using a fluorescence microscope. Blood vessels, proliferating cells or apoptotic cells were quantified from five random areas in each section. To evaluate the potential toxic effects of PMSCs on the host, tissues of the heart, liver, spleen, lung, kidney and brain were fixed in 4% neutral paraform. Next, 3- to 5-μm sections were stained with hematoxylin and eosin (H&E).
Statistical analysis
The results of the statistical analyses are presented as the mean ± standard deviation (SD) for comparison of individual time points, and differences between the groups were tested via the Kaplan-Meier method and an unpaired Student's t-test. Survival data were plotted on a Kaplan-Meier curve. All p-values were two-sided, and the statistical significance was defined as p < 0.05.
Results
Cell surface phenotype and Multipotent differentiation Potential of PMSCs
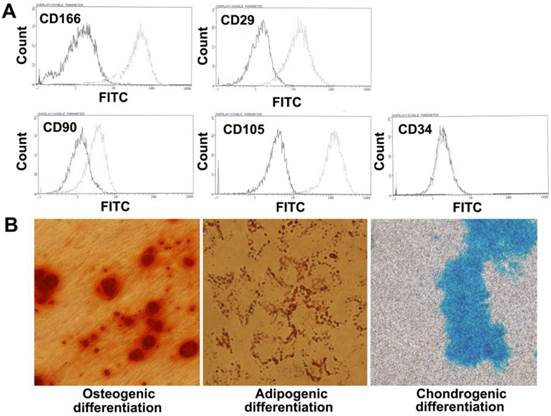
Flow cytometry analyses demonstrated that the PMSCs were positive for CD166, CD29, CD90, and CD105 and negative for hematopoietic markers CD34, which was consistent with the characteristics of BMSCs.
To evaluate the differentiation potential of PMSCs, cells were cultured with induction medium as previously described. As shown in Fig. 1, mineral depositions were detected using Alizarin red S staining for osteogenesis, accumulated triacylglycerol was detected using Oil red O staining for adipogenesis, and cartilage-specific proteoglycans were detected using Alcian blue staining for chondrogenesis. Taken together, these data suggested that PMSCs have multipotent differentiation potential.
Phenotype characteristics of PMSCs. A. Flow cytometry analysis of MSCs isolated from the placenta shows that the majority of cells are CD166+, CD29+, CD90+, CD105+, CD34-. The solid line denotes unstained control cells and dotted line denotes isolated PMSCs. B. Multipotent differentiation of PMSCs. Osteogenic, adipogenic and chondrogenic differentiation and histological staining of human placenta-derived MSCs using Alizarin red S, Oil red O and Alcian blue, respectively (magnification×200).
Expression of Ad-Endo in vitro
To improve the transduction efficiency of PMSCs, Lipofectamine 2000 was used to mediate adenoviral transduction. As shown in Fig. 2A, a very significant increase in the expression of EGFP in PMSCs was observed when the cells were transfected with Lipofectamine 2000, and a high-level viral titer resulted in enhanced expression of EGFP. Furthermore, flow cytometry was used to determine the transduction efficiency, in which 29.7% of PMSCs cells expressed EGFP at an MOI of 1500. This value was increased to 66.3% at an MOI of 3000 using Lipofectamine 2000 (Fig. 2B). These data were consistent with the results obtained from microscopic quantification. On the basis of these results, we selected an MOI of 3000 mixed with Lipofectamine 2000 at a ratio of 2.5×108 vp/µg for subsequent experiments.
Transfection efficiency of Ad-EGFP and the expression of endostatin, as determined using western blotting analyses. A. Transfection efficiency of Ad-EGFP was examined using fluorescence images (magnification ×40). B. Flow cytometry was performed to evaluate the transfection efficiency of Ad-EGFP. C. Expression of endostatin in PMSCs was detected using western blotting analyses, and β-actin was used as a loading control.
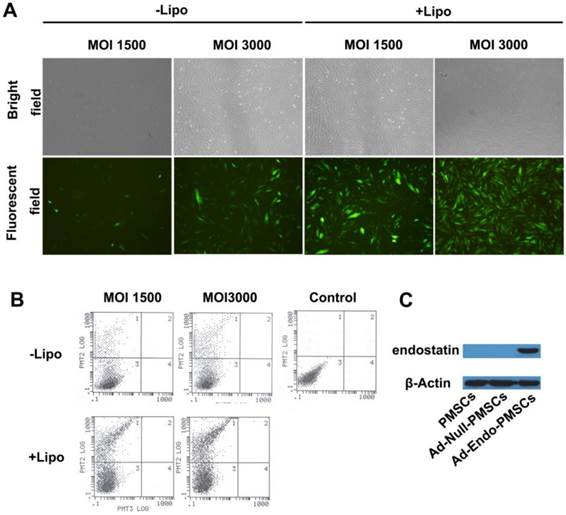
Western blotting analyses were performed to demonstrate the expression of Ad-Endo in PMSCs. As shown in Fig. 2C, a distinct band at approximately 20 kDa was detected in the CM from the Ad-Endo-transfected PMSCs but not in those from the untransfected PMSCs or Ad-Null-transfected PMSCs. These results indicated that endostatin was successfully synthesized and processed in vitro.
Migratory ability of Ad-Endo-PMSCs to CT26 in vitro
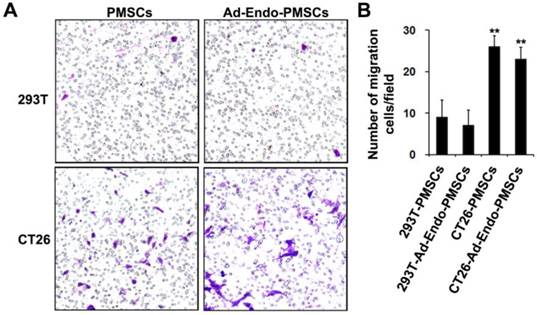
The migration ability of PMSCs towards tumors has been widely reported for tumors such as breast carcinoma, melanoma, Kaposi's sarcoma, glioma and prostate cancer. To investigate whether PMSCs have the ability to migrate toward CT26 cells, an in vitro transwell migration assay was performed. As shown in Fig. 3, there was a significant (p < 0.01) increase in the migration of PMSCs towards CT26 cells (26 ± 2.7) compared with 293T (9 ± 4.2). Moreover, the migratory ability of Ad-Endo-PMSCs towards CT26 cells (23 ± 2.9) occurred in a similar manner as that of untransfected PMSCs. These results strongly supported that CT26 cells were capable of stimulating the migration of PMSCs and that the migratory ability of PMSCs was not affected by Ad-Endo modification.
Migration ability of Ad-Endo-PMSCs to CT26 cells was examined using a transwell assay. A. The number of PMSCs targeted to CT26 showed a significant increase compared to cells targeted to 293T groups (magnification ×40). There was no significant difference between PMSCs and Ad-Endo-PMSCs in terms of their ability to target tumor cells. B. Average number of cells migrated in the transwell assay. The results were expressed as the mean ± S.D. in five different fields from three independent experiments (**p<0.01).
Tube formation assay
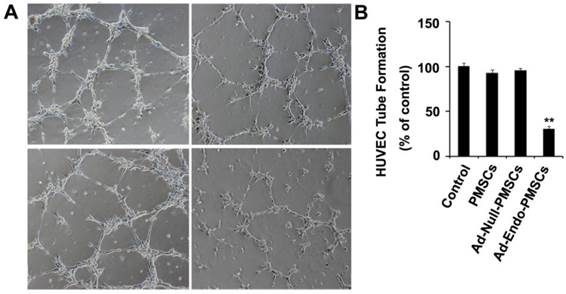
The bioactivity of endostatin expressed by Ad-Endo-PMSCs was confirmed in vitro using a tube formation assay. As shown in Fig. 4, HUVECs rapidly aligned and formed capillary-like structures when seeded onto matrigel (100 ± 15.3), and the CM derived from PMSCs (92.8 ± 13.7) or Ad-Null-PMSCs (95.6 ± 21.7) did not affect tube formation. However, there was significant inhibition of endothelial cell tube formation when HUVECs were treated with CM obtained from Ad-Endo-PMSCs (30.4 ± 7.4) (p <0.01). This can be attributed to the expression of Ad-Endo in PMSCs.
Ad-Endo-PMSCs inhibited tube formation of HUVECs. HUVECs cultured in EBM-2 medium treated with conditioned media from PMSCs, Ad-Null-PMSCs, or Ad-Endo-PMSCs for 6 h, respectively. Fresh DMEM-LG was used as a negative control. B. Quantitative summary of tube formation was performed by manual quantification of closed tubes in five different areas. All experiments were repeated three times (**p <0.05).
Ad-Endo-PMSCs significantly inhibited the peritoneal dissemination and growth of colorectal cancer in vivo
To examine the therapeutic efficiency of Ad-Endo-PMSCs, the CT26 CRPC model was used. As was shown in Table 1, a sharp decrease in metastatic foci numbers was observed after intraperitoneal infusion of Ad-Endo-PMSCs. Systemic therapy with Ad-Endo-PMSCs resulted in apparent tumor inhibition versus Controls, PMSCs or Ad-Null-PMSCs (p < 0.05). Furthermore, a decrease in the size of tumor nodes and an increase in the tumor-free rate were observed in the Ad-Endo-PMSCs group compared to other groups (Fig. 5A).
Anti-metastasis induced by Ad-Endo-PMSCs in vivo
| Groups | Mean number of tumor nodes | Tumor free rate% |
|---|---|---|
| Control | 104.4±21.7 | 0 |
| PMSCs | 69.6±29.5 | 10.46 |
| Ad-Null-PMSCs | 75.2±17.8 | 14.32 |
| Ad-Endo-PMSCs | 8.2±3.79* | 56.3 |
*indicates p< 0.05
Tumor suppression and survival advantage in Ad-Endo-PMSC-treated mice. A. Images of representative CT26 tumor-bearing mice after intraperitoneal treatment. Ad-Endo-PMSCs effectively inhibited the peritoneal dissemination and growth of colorectal cancer. B. Significant increases in survival rates and prolonged survival times were observed in Ad-Endo-PMSC-treated mice compared to control groups. p<0.05, N=10 per group.
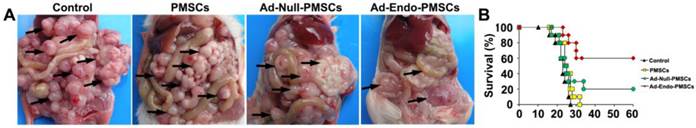
In the CT26 CRPC model, control animals receiving NaCl or PMSCs treatment survived 21 and 22.5 days on average, respectively, and the survival rate of mice in the Ad-Null-PMSCs group was 20%. In contrast, therapy with Ad-Endo-PMSCs resulted in a significant 3-fold increase in lifespan compared to Ad-Null-PMSCs (Fig. 5B). This result demonstrated that Ad-Endo-PMSCs have a significant and prolonged inhibitory effect on the tumor growth of CT26 in vivo.
Inhibition of tumor angiogenesis and cell proliferation in vivo
To determine the effects on tumor-induced angiogenesis, frozen tumor sections were stained using the CD31 antibody for immunofluorescence. According to Fig. 6A, micro-vessel densities (MVDs) of tumor tissues from Ad-Endo-PMSCs-treated mice (19 ± 4) showed a decreased number of CD31-positive vessels, which was significantly lower than the mice treated with NaCl (72 ± 7.1), PMSCs (58 ± 3.2) or Ad-Null-PMSCs (64 ± 7.8) (p < 0.05).
Ad-Endo-PMSCs inhibited tumor angiogenesis and cell proliferation and induced tumor apoptosis in vivo (1, Control; 2, PMSCs; 3, Ad-Null-PMSCs; 4, Ad-Endo-PMSCs, magnification ×200). A. Immunofluorescence assay of CD31. The Ad-Endo-PMSCs group showed a significant decrease in vessel density compared to the control groups. Vessel density was determined by quantification of the number of the vessels from five high-power fields. Values were expressed as the mean ± SD (*p < 0.05). B. Inhibition of cell proliferation within the tumor estimated using Ki67 immunohistochemical analysis. Tumors of the Ad-Endo-PMSCs group showed a decreased number of Ki67-positive cells compared to the other groups, particularly the NS groups. The proliferation index was determined by counting the number of the Ki67-positive cells from five high-power fields. Values were expressed as the mean ± SD (5 high power fields/slide, *p< 0.05). C. Apoptotic cells from CT26 tumors were detected using the TUNEL assay. The apoptotic index was calculated by dividing the number of TUNEL-positive cells by the total number of cells. Values were expressed as the mean ± SD (five high-power fields/slide,*p< 0.05).
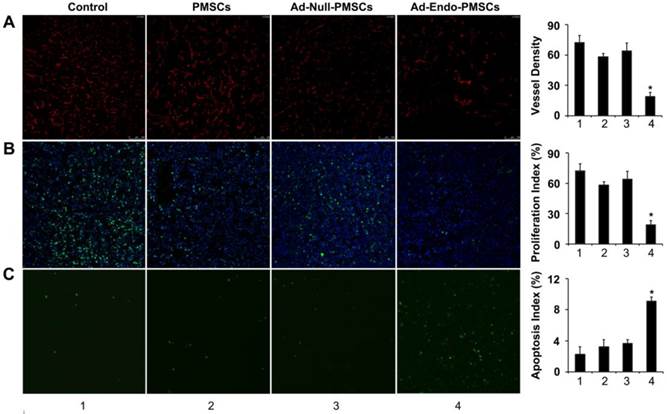
We also analyzed the effect induced by Ad-Endo-PMSCs on tumoral cell proliferation using Ki-67 staining. In all cases, we observed lower Ki-67 levels in tumors obtained from mice treated with Ad-Endo-PMSCs compared to the other groups, indicating a decrease in cell proliferation. These data suggested that the administration of Ad-Endo-PMSCs could effectively inhibit angiogenesis and colon tumor cell proliferation (Fig. 6B).
Increase in apoptosis in the Ad-Endo-PMSCs group
Apoptotic cells in tumors were determined using the TUNEL assay. As shown in Fig. 6C, the number of apoptotic cells in the Ad-Endo-PMSCs group was significantly higher compared to that of the other groups. The apoptotic index in Ad-Endo-PMSCs was 9.13 ± 0.49%, versus 2.27 ± 0.95% in the Control group (p < 0.01), 3.23 ± 0.89% in the PMSCs group (p < 0.05) and 3.63 ± 0.49 % in the Ad-Null-PMSCs group (p < 0.05).
Discussion
Angiogenesis plays an important role in the process of solid tumor growth and metastasis. Peritoneal tumors are malignant solid tumors that are also angiogenesis dependent [29]. Evidence from preclinical and clinical studies indicates that endostatin plays a central role in inhibiting angiogenesis and metastasis of primary solid malignancies [30, 31]. However, due to the high cost of producing large quantities of proteins and the short serum half-time of endostatin, it is necessary to exploit a long-term, cost-effective, and safe bio-delivery platform.
Gene therapy has been proposed as a potential treatment modality for advanced colorectal cancer, but it has been limited by insufficient gene delivery to tumor sites.
Viruses offer an ideal means of gene transfer, with adenoviruses being the most widely used due to their high gene transfer efficiency and wide host range [32, 33]. However, the transduction efficiency of adenoviruses is closely correlated with the cell surface density of the coxsackie-adenovirus receptor (CAR), and the lack of high-affinity CAR expression is a potential cause of intrinsic resistance of tumor cells to this type of therapy. Unfortunately, the expression of CAR is highly variable and often low in primary cancer cells, which limits the use of adenoviral vectors (Ad) for gene therapy. In addition, the utility of adenoviral vectors for cancer therapy is limited due to their lack of specificity, as CAR is also expressed on normal cells. Previous studies have documented that adenovirus preferentially accumulates on cells in the liver and other organs via CAR. Furthermore, it remains a major challenge to maintain prolonged gene expression and to overcome the immune response against the virus itself [34]. Thus, new delivery tools are needed to ensure that anti-tumor genes are specifically expressed in the tumor site locally and constantly.
Since bone-forming progenitor cells were first isolated from rat marrow by Friedenstein and Petrakova in 1966 [35], MSCs have been widely used in pre-clinical and clinical therapy due to their apparent ability to home to the tumor site and its long-term gene expression without immune system surveillance [36, 37]. Thus far, different anti-tumor genes have been delivered to various carcinoma models using MSCs, and Kim SM et al. reported that MSCs engineered by TRAIL could efficiently inhibit intracranial glioma growth [38]; Studeny M et al. demonstrated that MSCs with forced expression of IFN-beta could produce biological agents locally at tumor sites and inhibited the growth of malignant cells [39]. However, bone marrow is still the most common source of MSCs for scientific and clinical purposes. However, there are several limitations and disadvantages in the use of BMSCs, such as the decreased expansion and differentiation potential with age. In addition, the invasive procedure for the collection of bone marrow and the low yield of MSCs make BMSCs impractical for therapeutic use. Thus, alternative sources of MSCs are in urgent demand. MSCs have been successfully isolated from many sources, including adipose tissue, umbilical cord blood, placenta [20], peripheral blood, connective tissues of the dermis and skeletal muscle [40]. Due to the natural MSCs surface phenotype, differentiation potential, immunosuppressive properties and safe peculiarity, placenta is thought to serve as an attractive reservoir of MSCs for clinical investigation and application [21, 22].
In this study, MSCs were isolated from maternal placental tissue following healthy full-term pregnancy and were dissociated into single cells or fluffy grape-like cell clusters using collagenase. According to the adherent growth characteristics of PMSCs, we used low-sugar low-serum medium for PMSC selection. Using this method, PMSCs maintained strong cell viability for 10 generations. In previous studies, we demonstrated that PMSCs positively expressed CD29, CD44, CD73, CD90, CD105, and CD166 and negatively expressed the hematopoietic stem cell surface antigen CD34, CD45, which was consistent with the criterion of MSCs. In the present studies, we confirmed that PMSCs have multipotent differentiation potential and can be differentiated into bone cells or cartilage cells or can undergo adipocyte differentiation under specific permissive conditions.
The effect of MSCs on the growth of tumors is still unclear. Several publications have reported the tumor-promoting properties of MSCs by participating in tumor stroma formation [41] or secreting tumor growth factors to create a metastasis-promoting microenvironment [42, 43]. In contrast to these results, Lu YR et al. [44] investigated the effects of MSCs on lymphoma, which indicated that MSCs could induce apoptosis and G0/G1 phase arrest of cancer cells through the up-regulation of p21 and caspase3; Chanda D et al. [45] reported that MSCs could inhibit prostate cancer progression by producing woven bone around the growing tumor cells. There is no obvious effect of PMSCs on tumor growth in our study, but the normalization of abnormal tumor vasculature was observed (Fig. 6A). Several studies have shown that the normalization of tumor vasculature can promote the delivery of drugs to tumors and improve the outcome of radiation therapy [46], which may be a potential mechanism of tumor growth inhibition by gene-modified MSCs. However, the underlying mechanisms require further study.
Cancers are sometimes considered 'wounds that never heal' because they can generate mobile MSCs by secreting chemokines and cytokines. Until recently, the recruitment of MSCs has been investigated in a variety of tumors, such as ovarian cancer, breast cancer, glioma, lung cancer, melanoma, osteosarcoma, among other cancers. As the targeting ability of MSCs directly affects their utility for targeting therapy, the enhancement of the targeting ability of MSCs will improve their application in clinical practice. In this study, we tested the targeting ability of PMSCs to CT26 cells in vitro and found that the modification of PMSCs by Ad-Endo did not affect their targeting ability.
In a preliminary study, we showed that human PMSCs can serve as cellular vehicles for targeting cancer. It has been demonstrated that Ad-Endo, which is transported by PMSCs, could improve anti-tumor effects and prolong the survival period in the CT26 CRPC model compared with Ad-Null-PMSCs. Thus, the potential mechanism of tumor growth inhibition may be related to PMSCs, which efficiently deliver genes to the target cells and protect Ad-Endo from being degraded by the immune system, thereby prolonging gene expression. Due to the advantages of PMSCs, the use of PMSCs during treatment can effectively prevent the serious adverse effects caused by repeated administration at a high dose.
In conclusion, our studies demonstrated that Ad-Endo-modified PMSCs could be used as a therapeutic approach in anti-angiogenesis gene therapy of colorectal cancer. Thus, the modified PMSCs might be a promising clinical application in future research.
Acknowledgements
The study is supported by the National High Technology Research and Development Program of China (863 Program) (2012AA020807), the National Science and Technology Major Projects for "Major New Drugs Innovation and Development" (2011ZX09102-101-03) and Program for New Century Excellent Talents in University (NCET) (NCET-09-0575).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Nie B, Shen Z, Wen JB, Wong OG, Hsueh WD, Huo LF. et al. AAV-HGFK1 and Ad-p53 cocktail therapy prolongs survival of mice with colon cancer. Molecular cancer therapeutics. 2008;7:2855-65 doi:10.1158/1535-7163.MCT-08-0366
2. Jayne DG, Fook S, Loi C, Seow-Choen F. Peritoneal carcinomatosis from colorectal cancer. The British journal of surgery. 2002;89:1545-50 doi:10.1046/j.1365-2168.2002.02274.x
3. Wang Y, Gong C, Yang L, Wu Q, Shi S, Shi H. et al. 5-FU-hydrogel inhibits colorectal peritoneal carcinomatosis and tumor growth in mice. BMC cancer. 2010;10:402. doi:10.1186/1471-2407-10-402
4. Ribatti D, Vacca A, Dammacco F. The role of the vascular phase in solid tumor growth: a historical review. Neoplasia. 1999;1:293-302
5. Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L. et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nature medicine. 2001;7:1194-201 doi:10.1038/nm1101-1194
6. Folkman J, Watson K, Ingber D, Hanahan D. Induction of Angiogenesis during the Transition from Hyperplasia to Neoplasia. Nature. 1989;339:58-61
7. Sakimoto I, Ohta K, Yamazaki T, Ohtani S, Sahara H, Sugawara F. et al. Alpha-sulfoquinovosylmonoacylglycerol is a novel potent radiosensitizer targeting tumor angiogenesis. Cancer research. 2006;66:2287-95 doi:10.1158/0008-5472.CAN-05-2209
8. Tandle A, Blazer DG 3rd, Libutti SK. Antiangiogenic gene therapy of cancer: recent developments. Journal of translational medicine. 2004;2:22. doi:10.1186/1479-5876-2-22
9. Abdollahi A, Hahnfeldt P, Maercker C, Grone HJ, Debus J, Ansorge W. et al. Endostatin's antiangiogenic signaling network. Molecular cell. 2004;13:649-63
10. Tjin Tham Sjin RM, Satchi-Fainaro R, Birsner AE, Ramanujam VM, Folkman J, Javaherian K. A 27-amino-acid synthetic peptide corresponding to the NH2-terminal zinc-binding domain of endostatin is responsible for its antitumor activity. Cancer research. 2005;65:3656-63 doi:10.1158/0008-5472.CAN-04-1833
11. Peng F, Xu Z, Wang J, Chen Y, Li Q, Zuo Y. et al. Recombinant human endostatin normalizes tumor vasculature and enhances radiation response in xenografted human nasopharyngeal carcinoma models. PloS one. 2012;7:e34646. doi:10.1371/journal.pone.0034646
12. Cho HM, Rosenblatt JD, Kang YS, Iruela-Arispe ML, Morrison SL, Penichet ML. et al. Enhanced inhibition of murine tumor and human breast tumor xenografts using targeted delivery of an antibody-endostatin fusion protein. Molecular cancer therapeutics. 2005;4:956-67 doi:10.1158/1535-7163.MCT-04-0321
13. Greco SJ, Rameshwar P. Enhancing effect of IL-1alpha on neurogenesis from adult human mesenchymal stem cells: implication for inflammatory mediators in regenerative medicine. J Immunol. 2007;179:3342-50
14. Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-kappaB signaling in resident macrophages. Blood. 2011;118:330-8 doi:10.1182/blood-2010-12-327353
15. Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Molecular cancer therapeutics. 2006;5:755-66 doi:10.1158/1535-7163.MCT-05-0334
16. Nakamura K, Ito Y, Kawano Y, Kurozumi K, Kobune M, Tsuda H. et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene therapy. 2004;11:1155-64 doi:10.1038/sj.gt.3302276
17. Nauta AJ, Kruisselbrink AB, Lurvink E, Willemze R, Fibbe WE. Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J Immunol. 2006;177:2080-7
18. Ryan JM, Barry FP, Murphy JM, Mahon BP. Mesenchymal stem cells avoid allogeneic rejection. J Inflamm (Lond). 2005;2:8. doi:10.1186/1476-9255-2-8
19. Kemp KC, Hows J, Donaldson C. Bone marrow-derived mesenchymal stem cells. Leukemia & lymphoma. 2005;46:1531-44 doi:10.1080/10428190500215076
20. Deak E, Seifried E, Henschler R. Homing pathways of mesenchymal stromal cells (MSCs) and their role in clinical applications. International reviews of immunology. 2010;29:514-29 doi:10.3109/08830185.2010.498931
21. Miao Z, Jin J, Chen L, Zhu J, Huang W, Zhao J. et al. Isolation of mesenchymal stem cells from human placenta: comparison with human bone marrow mesenchymal stem cells. Cell biology international. 2006;30:681-7 doi:10.1016/j.cellbi.2006.03.009
22. In 't Anker PS, Scherjon SA, Kleijburg-van der Keur C, de Groot-Swings GM, Claas FH, Fibbe WE. et al. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells. 2004;22:1338-45 doi:10.1634/stemcells.2004-0058
23. Zhang D, Tong A, Zhou L, Fang F, Guo G. Osteogenic differentiation of human placenta-derived mesenchymal stem cells (PMSCs) on electrospun nanofiber meshes. Cytotechnology. 2012 doi:10.1007/s10616-012-9450-5
24. Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R. et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309:1074-8 doi:10.1126/science.1110955
25. Huang YC, Yang ZM, Chen XH, Tan MY, Wang J, Li XQ. et al. Isolation of mesenchymal stem cells from human placental decidua basalis and resistance to hypoxia and serum deprivation. Stem cell reviews. 2009;5:247-55 doi:10.1007/s12015-009-9069-x
26. Murdoch AD, Grady LM, Ablett MP, Katopodi T, Meadows RS, Hardingham TE. Chondrogenic differentiation of human bone marrow stem cells in transwell cultures: generation of scaffold-free cartilage. Stem Cells. 2007;25:2786-96 doi:10.1634/stemcells.2007-0374
27. Menon LG, Picinich S, Koneru R, Gao H, Lin SY, Koneru M. et al. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells. 2007;25:520-8 doi:10.1634/stemcells.2006-0257
28. Tsai AC, Pan SL, Sun HL, Wang CY, Peng CY, Wang SW. et al. CHM-1, a new vascular targeting agent, induces apoptosis of human umbilical vein endothelial cells via p53-mediated death receptor 5 up-regulation. The Journal of biological chemistry. 2010;285:5497-506 doi:10.1074/jbc.M109.036277
29. Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell stem cell. 2009;4:206-16 doi:10.1016/j.stem.2009.02.001
30. Brideau G, Makinen MJ, Elamaa H, Tu H, Nilsson G, Alitalo K. et al. Endostatin overexpression inhibits lymphangiogenesis and lymph node metastasis in mice. Cancer research. 2007;67:11528-35 doi:10.1158/0008-5472.CAN-07-1458
31. Noro T, Miyake K, Suzuki-Miyake N, Igarashi T, Uchida E, Misawa T. et al. Adeno-associated viral vector-mediated expression of endostatin inhibits tumor growth and metastasis in an orthotropic pancreatic cancer model in hamsters. Cancer research. 2004;64:7486-90 doi:10.1158/0008-5472.CAN-03-1296
32. Holst PJ, Orskov C, Thomsen AR, Christensen JP. Quality of the transgene-specific CD8+ T cell response induced by adenoviral vector immunization is critically influenced by virus dose and route of vaccination. J Immunol. 2010;184:4431-9 doi:10.4049/jimmunol.0900537
33. Tatsis N, Ertl HC. Adenoviruses as vaccine vectors. Molecular therapy: the journal of the American Society of Gene Therapy. 2004;10:616-29 doi:10.1016/j.ymthe.2004.07.013
34. Thaci B, Ulasov IV, Wainwright DA, Lesniak MS. The challenge for gene therapy: innate immune response to adenoviruses. Oncotarget. 2011;2:113-21
35. Friedenstein AJ, Piatetzky S II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. Journal of embryology and experimental morphology. 1966;16:381-90
36. Stappenbeck TS, Miyoshi H. The role of stromal stem cells in tissue regeneration and wound repair. Science. 2009;324:1666-9 doi:10.1126/science.1172687
37. Chamberlain G, Fox J, Ashton B, Middleton J. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25:2739-49 doi:10.1634/stemcells.2007-0197
38. Kim SM, Lim JY, Park SI, Jeong CH, Oh JH, Jeong M. et al. Gene therapy using TRAIL-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer research. 2008;68:9614-23 doi:10.1158/0008-5472.CAN-08-0451
39. Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer research. 2002;62:3603-8
40. Wagner W, Wein F, Seckinger A, Frankhauser M, Wirkner U, Krause U. et al. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Experimental hematology. 2005;33:1402-16 doi:10.1016/j.exphem.2005.07.003
41. Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN. et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. Journal of the National Cancer Institute. 2004;96:1593-603 doi:10.1093/jnci/djh299
42. Mi Z, Bhattacharya SD, Kim VM, Guo H, Talbot LJ, Kuo PC. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis. 2011;32:477-87 doi:10.1093/carcin/bgr009
43. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557-63 doi:10.1038/nature06188
44. Lu YR, Yuan Y, Wang XJ, Wei LL, Chen YN, Cong C. et al. The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer biology & therapy. 2008;7:245-51
45. Chanda D, Isayeva T, Kumar S, Hensel JA, Sawant A, Ramaswamy G. et al. Therapeutic potential of adult bone marrow-derived mesenchymal stem cells in prostate cancer bone metastasis. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:7175-85 doi:10.1158/1078-0432.CCR-09-1938
46. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58-62 doi:10.1126/science.1104819
Author contact
![]() Corresponding author: Yan Luo yanluo300com. Li Yang yl.tracy73com; Tel: + 86-13730830347; Fax: + 86-28-85164060
Corresponding author: Yan Luo yanluo300com. Li Yang yl.tracy73com; Tel: + 86-13730830347; Fax: + 86-28-85164060