Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(7):742-747. doi:10.7150/ijms.7167 This issue Cite
Short Research Communication
Cyanidin 3-O-Glucoside Reduces Helicobacter pylori VacA-Induced Cell Death of Gastric KATO III Cells through Inhibition of the SecA Pathway
Sa-Hyun Kim1*, Hyunjun Woo2*, Min Park2, Ki-Jong Rhee2, Cheol Moon1, Dongsup Lee3, Woo Duck Seo4 ![]() , Jong Bae Kim2
, Jong Bae Kim2 ![]()
1. Department of Clinical Laboratory Science, Semyung University, Jaecheon 390-711, Republic of Korea;
2. Department of Biomedical Laboratory Science, College of Health Science, Yonsei University, Wonju 220-710, Republic of Korea;
3. Department of Clinical Laboratory Science, Hyegeon College, Hongseong 350-702, Republic of Korea;
4. Department of Functional Crops, National Institute of Crop Science, Rural Development Administration, Miryang 627-803, Republic of Korea.
* These investigators contributed equally to this study.
Received 2013-7-15; Accepted 2014-4-21; Published 2014-5-14
Abstract
Two key virulence factors of Helicobacter pylori are the secreted virulent proteins of vacuolating toxin A (VacA) and cytotoxin associated protein A (CagA) which lead to damages of gastric epithelial cells. We previously identified that the cyanidin 3-O-glucoside (C3G) inhibits the secretion of both VacA and CagA. In the current report, we show that C3G inhibits VacA secretion in a dose-dependent manner by inhibiting secretion system subunit protein A (SecA) synthesis. As SecA is involved in translocation of bacterial proteins, we predicted that inhibition of the SecA pathway by C3G should decrease H. pylori-induced cell death. To test this hypothesis, the human gastric cell line KATO III cells were co-cultured with H. pylori 60190 (VacA+/CagA+) and C3G. We found that C3G treatment caused a decrease in activation of the pro-apoptotic proteins caspase-3/-8 in H. pylori-infected cells leading to a decrease in cell death. Our data suggest that consumption of foods containing anthocyanin may be beneficial in reducing cell damage due to H. pylori infection.
Keywords: H. pylori, cyanidin 3-O-glucoside, VacA secretion
Introduction
During H. pylori pathogenesis, many virulence factors such as toxins, adhesins, proteases are involved in infecting gastric cells. Among them, vacuolating cytotoxin A (VacA) and cytotoxin associated protein A (CagA) are two important virulence factors contributing to cellular damage. VacA functions as an intracellular-acting protein exotoxin1, and causes cellular apoptosis1, 2, gastric epithelial cell damage by inducing vacuoles3-5. CagA also induces cellular apoptosis6, interleukin-8 secretion7, 8 and cancer development via multiple pathways9-11. Patients harboring a H. pylori strain containing the Eastern type of the cagA gene have a much higher risk developing gastric ulceration and stomach cancer12-14. Although the mechanism by which these two virulent proteins are secreted is still under intense investigation, VacA secretion is regulated by the type Va secretion system (T5aSS) which is SecA-dependent3, 17 whereas CagA secretion appears to occur through the T4SS which is SecA-independent15, 16. Sec proteins are associated with Sec protein in T5aSS3, 15, 18. Of these Sec proteins secretion system subunit protein A (SecA) is an intracellular protein that is involved in translocation of bacterial proteins out of the bacterial plasma membrane19.
There is a positive correlation between consumption of anthocyanin-containing foods and disease prevention20,21. The potential disease-preventive actions of anthocyanins include antibacterial effects22,23, inhibitory effects on the development of peptic ulcers and gastric cancer10,14,24, and reduction in interleukin-8 secretion7. However, in many studies of anthocyanins, crude extracts were used and thus the beneficial effects attributed to anthocyanins are unclear22, 23. In a previous study, we demonstrated that cyanidin 3-O-glucoside (C3G) inhibited secretion of H. pylori virulent proteins21. In the current report, using a newly developed anti-SecA antibody, we found that C3G inhibited synthesis of SecA resulting in inhibition of VacA secretion. In addition, C3G decreased H. pylori-induced apoptosis of KATO III gastric cells.
Materials and Methods
Cyanidin 3-O-glucoside
C3G (Extrasynthese; Lyon, France) was dissolved in sterile dimethyl sulfoxide (DMSO; Sigma Aldrich, St. Louis, MO, USA) as a 50 mM stock solution and stored at -20°C prior to usage.
Bacterial strain and culture
Helicobacter pylori reference strain 60190 (Eastern type: CagA+/VacA+) was purchased from American Type Cell Collection (ATCC; Manassas, VA, USA). Bacteria were maintained under microaerophilic conditions at 37°C on Brucella agar plates (Becton-Dickinson, Braintree, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Long Island, NY, USA). For experiments, H. pylori were cultured in Mueller-Hinton broth (Becton-Dickinson) containing 10% FBS under microaerophilic condition for 3~4 days at 37°C and 100% humidity.
Cell line and culture
KATO III (human gastric carcinoma line) was purchased from ATCC. KATO III cells were grown in Iscove's Modified Dulbecco's Medium (IMDM) with 20% FBS at 37°C and 5% CO2 concentration. Cells were seeded in 10-cm plates (5 × 106) 24 h before infection with H. pylori 60190 strain [200 multiplicity of infection (MOI)] and then treated with C3G for two days. Control groups were mock-treated with DMSO.
Antibodies
New Zealand White rabbits (8~10 wk) were purchased from Central Lab Animal Inc. (Seoul, Korea) and allowed to adapt to their new environment for two weeks before the first antigen inoculation. The anti-SecA polyclonal antiserum was generated by intravenous injection of 500 μg of a 22 mer peptide (GTERHESRRIDNQLRGRSGRQG; nucleotide 518-539) every week for a total of six weeks. After six weeks, blood was collected, allowed to clot at 4°C overnight and the serum was isolated after centrifugation. Pre-immune serum was harvested prior to immunization. Antibodies were purified using a protein A column and reactivity to SecA confirmed by ELISA (data not shown). Rabbit anti-H. pylori polyclonal antibody to whole H. pylori 60190 was generated as previously described21.
Western blot analysis
H. pylori were lysed in ice-cold RIPA lysis buffer (Millipore, Billerica, MA, USA) for 30 minutes on ice and sonicated for 2 minutes with 10 second intervals (Sonicator XL-2020, Heat Systems Ultrasonics, Pittsburgh, PA, USA). KATO III cells (5 × 106) were plated in 10-cm plates. Following infection, cells were washed with PBS, lysed with 150 μL of RIPA buffer on ice for 30 min. Lysates of culture dishes treated identically were pooled and clarified at 13,000 × g for 10 min at 4°C. Protein concentration was determined by the Lowry method and quantified using NanoQuant spectrophotometer (Infinite M200, TECAN, Männedorf, Switzerland). Protein extracts were resolved on 7.5, 10% or 4~15% gradient SDS-PAGE and then transferred to a nitrocellulose membrane (Millipore). Membranes were blocked with 5% skim milk for 30 minutes and then incubated with rabbit anti-VacA polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-Helicobacter pylori SecA polyclonal antibody (this study) or rabbit anti-Helicobacter pylori polyclonal antibody21. KATO III cell lysates were immunoblotted with rabbit anti-cleaved caspase-8 monoclonal antibody (Cell Signaling Technology, Danvers, MA, USA), rabbit anti-cleaved caspase-3 monoclonal antibody (Cell Signaling Technology), rabbit anti-cleaved PARP monoclonal antibody (Cell Signaling Technology) or rabbit anti-α-tubulin monoclonal antibody (Cell Signaling Technology). Appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) were used and protein bands were visualized using enhanced chemiluminescence and X-ray film. The bacterial supernatant was centrifuged at 3,000 rpm three times and filtered with 0.22 μm syringe filters to remove remained the last bacterium. The prepared supernatant was concentrated 10-fold using Centricon centrifugal filters (3 kDa cut-off) (Millipore) at 3,000 rpm for 1~2 hours at 4°C.
MTT assay
Cell viability was measured by a quantitative colorimetric assay with 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT). After treatment with C3G, cells were incubated with MTT reagent (final concentration 0.1 mg/ml) for 150 min at 37℃. The reaction was terminated by addition of DMSO. The amount of MTT formazon product was determined by measuring the absorbance at 560 nm using NanoQuant spectrophotometer (Infinite M200, TECAN). The change in cell viability was calculated as follows:
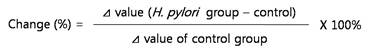
Annexin V and 7-AAD (7-Amino-Actinomycin) binding staining
The assay of Annexin V and 7-AAD (7-Amino-Actinomycin) binding staining was performed with an Annexin V-PE Apoptosis Detection Kit I according to the manufacturer's instructions (Becton-Dickinson). In brief, cells after H. pylori infection (200 MOI) were detached with 0.25% trypsin without EDTA, and then washed twice with cold PBS, centrifuged at 3000 rpm for 5 minutes. Cells were resuspended in 500 μL of 1× binding buffer at a concentration of 5 × 105 cells/mL, 5 μL Annexin V-PE and 5 μL 7-AAD were added. Cells were gently mixed and incubated for 10 minutes at 37°C in the dark. Transfer 400 μL of cell suspension to flow tubes. Stained cells were analyzed by FACS Caliber flow cytometry (Becton-Dickinson).
Results and Discussion
Secretion of VacA is inhibited by cyanidin 3-O-glucoside in a dose-dependent manner
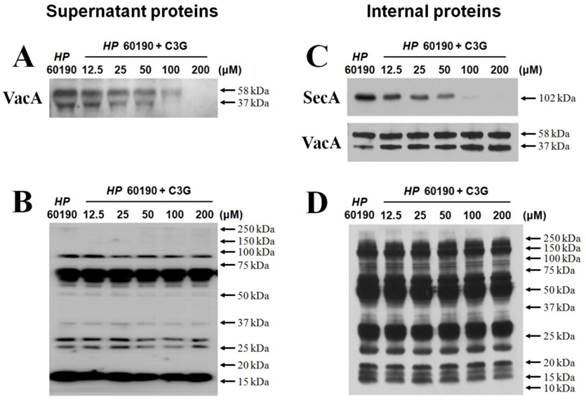
We previously showed that C3G inhibited the secretion of virulent proteins in H. pylori resulting in intracellular accumulation in periplasmic space21. In the current study, we conducted a dose response to determine the effective concentration of C3G. H. pylori 60190 were cultured with different concentrations of C3G for three days and the secreted VacA was assessed by Western blot analysis. We found that the levels of both secreted VacA was decreased when bacteria were cultured with C3G starting a 12.5 μM of C3G and this decrease was more pronounced proportionately to increases in C3G concentration (Figure 1A). Consistent with previous results21, the bacterial numbers were unchanged by C3G treatment at all concentrations compared to mock-treated bacteria suggesting that the decrease in secreted VacA was not due to a decrease in bacterial cell numbers (data not shown). The levels of total secreted proteins were similar at all concentrations of C3G used as determined by reactivity to rabbit anti-H. pylori polyclonal antibody showing that the decrease in VacA was specific to C3G treatment (Figure 1A and B). The pre-immune serum showed negligible detection to H. pylori secreted proteins (data not shown). The increased level of VacA was due to inhibition of secretion and not protein synthesis as intracellular levels of VacA did not decrease in response to C3G treatment with C3G (Figure 1C and D).
Effects of cyanidin 3-O-glucoside on secretion of H. pylori VacA and expression of SecA. H. pylori was cultured with various concentrations (0~100 μM) of C3G in Mueller-Hinton broth/10% FBS for 3 days and proteins assessed by Western blot. (A), secreted VacA in culture media. (B), secreted H. pylori proteins reacting with rabbit anti-H. pylori polyclonal antibody. (C), intracellular SecA and VacA. (D), intracellular H. pylori proteins reacting with rabbit anti-H. pylori polyclonal antibody. Representative image were obtained from five independent experiments.
SecA synthesis is inhibited by cyanidin 3-O-glucoside treatment in a dose-dependnet manner
In our previous report we showed that C3G inhibited secA transcription21. SecA is an intracellular protein that facilitates translocation of bacterial proteins out of the bacterial plasma membrane19. The decrease in secA transcription by C3G offers a mechanistic explanation as to why C3G induces intracellular accumulation of VacA. We reconfirmed that C3G specifically inhibits secA transcription by RT-PCR analysis (data not shown). In the current study, we developed SecA specific polyclonal antibodies and tested whether SecA protein levels were also decreased in response to C3G treatment. As expected, we found a dose-dependent decrease in SecA proteins levels in response to C3G treatment in H. pylori (Figure 1C, D). Taken together, our data shows that inhibition of VacA secretion in H. pylori strongly correlates with decreased SecA mRNA transcription and protein synthesis.
Cyanidin 3-O-glucoside alleviates H. pylori-induced apoptosis in the KATO III cell line
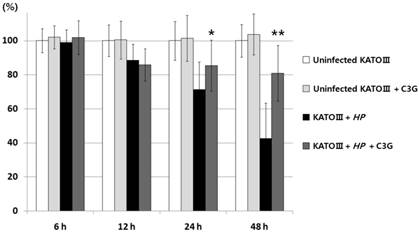
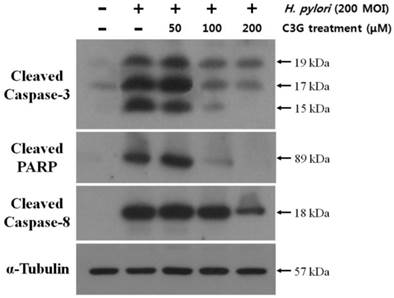
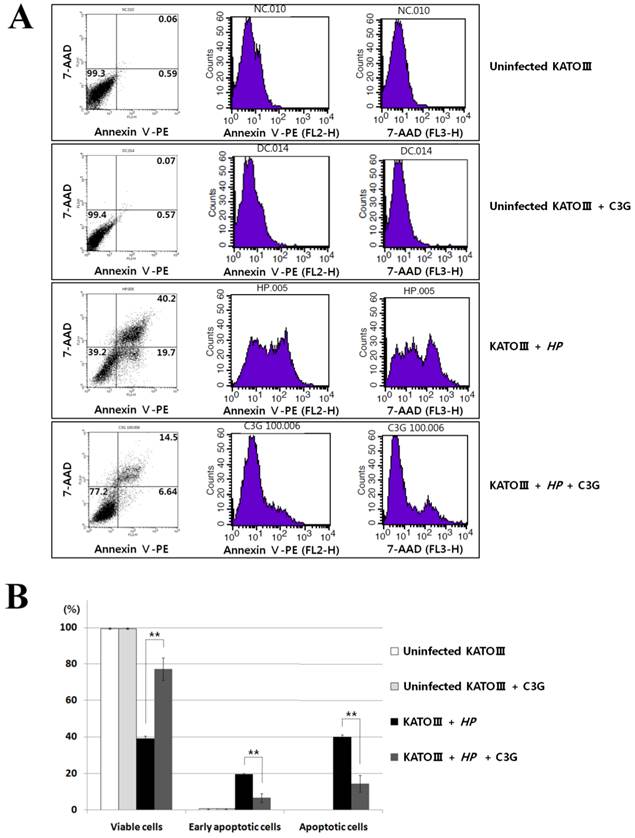
VacA causes apoptotic cell death of gastric cell lines. Since C3G inhibits VacA secretion, we predicted that C3G inhibits H. pylori infection-mediated cell death in gastric cells. To test this idea, the human gastric cancer cell line KATO III cells were infected with H. pylori (200 MOI) and treated with C3G (100 μM) for up to 48 hr. Whereas cells infected with H. pylori showed a 42% cell viability at 48 h compared to uninfected controls, the C3G treated group showed a 81% cell viability (p < 0.001, Figure 2). The decrease in cell death was likely due to a decrease in the apoptotic signaling pathway as cells co-cultured with C3G and H. pylori exhibited decreased levels of the pro-apoptotic molecules, cleaved caspase-3, -8 and cleaved PARP compared to non-treated controls (Figure 3). Then the assay of Annexin V and 7-AAD (7-Amino-Actinomycin) binding staining results was also showed reduced apoptosis in H. pylori-infected KATO III when cells were exposed to C3G (100 μM), whereas cell viability of infected cells (HP+ / C3G-) was not recovered compared to uninfected controls (HP- / C3G- or HP- / C3G+) and C3G-treated groups (HP+ / C3G+) as shown in Figure 4A, B (p < 0.001).
Effect of cyanidin 3-O-glucoside on gastric KATO Ⅲ cell death by H. pylori infection. Cell viability was measured by a quantitative colorimetric MTT assay. Human gastric cancer cell line, KATO III cells were infected with H. pylori (200 MOI) and treated with C3G (100 μM) for up to 48 hr. The C3G treated group showed a 81% cell viability, whereas cells infected with H. pylori showed a 42% cell viability at 48 h compared to uninfected controls (*: p < 0.01, **: p < 0.001).
Effects of cyanidin 3-O-glucoside treatment on apoptotic signal pathways in KATO Ⅲ cells. KATO III cells were infected with H. pylori (200 MOI) and exposed to various concentrations of C3G (0~200 μM) for 48 hr. The dose-dependent decreases in expressions of cleaved caspase-3, -8, and cleaved PARP by H. pylori infection (200 MOI) were shown significantly by treatment of C3G.
Effects of cyanidin 3-O-glucoside treatment on apoptosis in H. pylori-infected KATO Ⅲ cells. (A), The assay of Annexin V and 7-AAD (7-Amino-Actinomycin) binding staining was performed with the same batch cells on Figure 3. Data analysed with FACS flow cytometry (Becton-Dickinson). (B), C3G treatment showed reduced apoptosis in H. pylori-infected KATO III cells, whereas cell viability of infected cells (HP+ / C3G-) was not recovered compared to uninfected control (HP- / C3G- or HP- / C3G+) and C3G-treated groups (HP+ / C3G+).
VacA secretion occurs via T5aSS3, 15, and it is thought that SecA in H. pylori is bound to the inner membrane complex composed of the Sec-related family proteins SecD, SecF, SecG and SecY19. This protein complex regulates the secretions of various secreted virulence factors such as proteins, toxins and proteases. Our data suggest that C3G down-regulates the secretion of VacA in H. pylori via inhibition of SecA expression and synthesis. Following our prior report21, C3G decreased the secA transcription. If so SecA expression was suppressed by C3G as shown in this report, because VacA secretion is regulated by SecA protein which is bound to inner membrane of H. pylori3,15, VacA secretion might be down-regulated. Thus cell survival rate would be also increased through the inhibition of VacA secretion when H. pylori-infected cells were exposed to C3G. Given that black fruits and crops are an important source of C3G, consumption of these foods may be beneficial in reducing gastric inflammation or stomach cancer due to H. pylori infection.
It has been reported that CagA secretion occurs via the T4SS3, 16, 17 and that CagA has no SecA secretion signal for transport25. And many researchers in the past have clearly excluded secretion of CagA in the supernatant26 because CagA is a T4SS effector protein which is only injected into the cells11-13. In this study, the accumulation of intracellular CagA protein into the cell was found repeatedly (data now shown in Figure 1C). It is thought that the accumulation of CagA in bacterial cell might make reduced injection of it into the cells, however this study could not show the evidence about the inhibition of CagA secretion by C3G.
Acknowledgements
This work was carried out with the supported of the "Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ010528)”, Rural Development Administration, Republic of Korea.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kim IJ, Blanke SR. Remodeling the host environment: modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front Cell Infect Microbiol. 2012;2:37
2. Oldani A, Cormont M, Hofman V, Chiozzi V, Oregioni O, Canonici A, Sciullo A, Sommi P, Fabbri A, Ricci V, Boquet P. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009;5:e1000603
3. Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin Multifunctionality. Nat Rev Microbiol. 2005;3:320-332
4. Rassow J. Helicobacter pylori vacuolating toxin A and apoptosis. Cell Commun Signal. 2011;9:26
5. Wang F, Xia P, Wu F, Wang D, Wang W, Ward T, Liu Y, Aikhionbare F, Guo Z, Powell M, Liu B, Bi F, Shaw A, Zhu Z, Elmoselhi A, Fan D, Cover TL, Ding X, Yao X. Helicobacter pylori VacA disrupts apical membrane-cytoskeletal interactions in gastric parietal cells. J Biol Chem. 2008;283:26714-26725
6. Chattopadhyay R, Bhattacharyya A, Crowe SE. Dual Regulation by Apurinic/Apyrimidinic Endonuclease-1 Inhibits Gastric Epithelial Cell Apoptosis during Helicobacter pylori Infection. Cancer Res. 2010;70:2799-2808
7. Kumar Pachathundikandi S, Brandt S, Madassery J, Backert S. Induction of TLR-2 and TLR-5 expression by Helicobacter pylori switches cagPAI-dependent signalling leading to the secretion of IL-8 and TNF-α. PLoS ONE. 2011;6:e19614
8. Schneider S, Carra G, Sahin U, Hoy B, Rieder G, Wessler S. Complex cellular responses of Helicobacter pylori-colonized gastric adenocarcinoma cells. Infect Immun. 2011;79:2362-2371
9. Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, Wroblewski LE, Piazuelo MB, Correa P, Peek RM Jr. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379-387
10. Gao L, Michel A, Weck MN, Arndt V, Pawlita M, Brenner H. Helicobacter pylori infection and gastric cancer risk: evaluation of 15 H. pylori proteins determined by novel multiplex serology. Cancer Res. 2009;69:6164-6170
11. Keates S, Keates AC, Nath S, Peek RM Jr, Kelly CP. Transactivation of the epidermal growth factor receptor by cag+ Helicobacter pylori induces upregulation of the early growth response gene Egr-1 in gastric epithelial cells. Gut. 2005;54:1363-1369
12. An B, Lee G, Lim K, Moon BS, Kim JB. Identification of the repeated number of C and D regions of tyrosine phosphorylation motifs in Helicobacter pylori cagA using multiplex PCR. Microbiol Immunol. 2008;52:479-483
13. Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553-1566
14. Yang JJ, Cho LY, Ko KP, Shin A, Ma SH, Choi BY, Han DS, Song KS, Kim YS, Lee JY, Han BG, Chang SH, Shin HR, Kang D, Yoo KY, Park SK. Genetic susceptibility on CagA-interacting molecules and gene-environment interaction with phytoestrogens: a putative risk factor for gastric cancer. PLoS ONE. 2012;7:e31020
15. Boquet P, Ricci V. Intoxication strategy of Helicobacter pylori VacA toxin. Trends in Microbiology. 2012;20:165-174
16. Fronzes R, Christie PJ, Waksman G. The structural biology of type IV secretion systems. Nature Rev Microbiol. 2009;7:703-714
17. Shaffer CL, Gaddy JA, Loh JT, Johnson EM, Hill S, Hennig EE, McClain MS, McDonald WH, Cover TL. Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog. 2011;7:e1002237
18. Mobley HL, Mendz GL, Hazell SL. Helicobacter pylori: Physiology and Genetics. ASM Press. 2001:219-224
19. Whitehouse S, Gold VA, Robson A, Allen WJ, Sessions RB, Collinson I. Mobility of the SecA 2-helix-finger is not essential for polypeptide translocation via the SecYEG complex. J Cell Biol. 2012;199:919-929
20. Matsushima M, Suzuki T, Masui A, Kasai K, Kouchi T, Takagi A, Shirai T, Mine T. Growth inhibitory action of cranberry on Helicobacter pylori. J Gastroenterol Hepatol. 2008;23:S175-180
21. Kim SH, Park M, Woo H, Tharmalingam N, Lee G, Rhee KJ, Eom YB, Han SI, Seo WD, Kim JB. Inhibitory effects of anthocyanins on secretion of Helicobacter pylori CagA and VacA toxins. Int J Med Sci. 2012;9:838-842
22. Côté J, Caillet S, Doyon G, Sylvain JF, Lacroix M. Bioactive compounds in cranberries and their biological properties. Crit Rev Food Sci Nutr. 2010;50:666-679
23. Neto CC. Cranberry and its phytochemicals: a review of in vitro anticancer studies. J Nutr. 2007;137:186S-193S
24. Bagnoli F, Buti L, Tompkins L, Covacci A, Amieva MR. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc Natl Acad Sci USA. 2005;102:16339-16344
25. Ripoll-Rozada J, Peña A, Rivas S, Moro F, de la Cruz F, Cabezón E, Arechaga I. Regulation of the type IV secretion ATPase TrwD by magnesium: implications for the catalytic mechanism of the secretion ATPase superfamily. J Biol Chem. 2012;287:17408-17414
26. Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287(5457):1497-1500
Author contact
![]() Corresponding author: Jong Bae Kim, D.V.M., PhD. Department of Biomedical Laboratory Science, College of Health Sciences, Yonsei University, Wonju, Gangwon, 220-710, Republic of Korea. e-mail: kimjb70ac.kr. Phone: +82-33-760-2423 Or Woo Duck Seo, PhD. Department of Functional Crops, National Institute of Crop Science, Rural Development Administration, Miryang, Gyeongnam, 627-803, Republic of Korea. e-mail: swd2002kr Phone: +82-55-350-1166.
Corresponding author: Jong Bae Kim, D.V.M., PhD. Department of Biomedical Laboratory Science, College of Health Sciences, Yonsei University, Wonju, Gangwon, 220-710, Republic of Korea. e-mail: kimjb70ac.kr. Phone: +82-33-760-2423 Or Woo Duck Seo, PhD. Department of Functional Crops, National Institute of Crop Science, Rural Development Administration, Miryang, Gyeongnam, 627-803, Republic of Korea. e-mail: swd2002kr Phone: +82-55-350-1166.