Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(4):373-380. doi:10.7150/ijms.7802 This issue Cite
Research Paper
Endothelin-1 Promotes Cardiomyocyte Terminal Differentiation in the Developing Heart via Heightened DNA Methylation
Alexandra Paradis1, Daliao Xiao1, Jianjun Zhou1,2, Lubo Zhang1 ![]()
1. Center for Perinatal Biology, Division of Pharmacology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, California 92350
2. Department of Obstetrics and Gynecology, Nanjing Drum Tower Hospital, Nanjing University Medical School, Nanjing 210008, China
Received 2013-10-3; Accepted 2013-12-17; Published 2014-2-20
Abstract
Aims: Hypoxia is a major stress on fetal development and leads to induction of endothelin-1 (ET-1) expression. We tested the hypothesis that ET-1 stimulates the terminal differentiation of cardiomyocytes from mononucleate to binucleate in the developing heart.
Methods and results: Hypoxia (10.5% O2) treatment of pregnant rats from day 15 to day 21 resulted in a significant increase in prepro-ET-1 mRNA expression in fetal hearts. ET-1 ex vivo treatment of fetal rat cardiomyocytes increased percent binucleate cells and decreased Ki-67 expression, a marker for proliferation, under both control and hypoxic conditions. Hypoxia alone decreased Ki-67 expression and in conjunction with ET-1 treatment decreased cardiomyocyte size. PD145065, a non-selective ET-receptor antagonist, blocked the changes in binucleation and proliferation caused by ET-1. DNA methylation in fetal cardiomyocytes was significantly increased with ET-1 treatment, which was blocked by 5-aza-2'-deoxycytidine, a DNA methylation inhibitor. In addition, 5-aza-2'-deoxycytidine treatment abrogated the increase in binucleation and decrease in proliferation induced by ET-1.
Conclusions: Hypoxic stress and synthesis of ET-1 increases DNA methylation and promotes terminal differentiation of cardiomyocytes in the developing heart. This premature exit of the cell cycle may lead to a reduced cardiomyocyte endowment in the heart and have a negative impact on cardiac function.
Keywords: Endothelin-1, Hypoxia, Heart, Fetal development, Epigenetic
Introduction
Heart disease is the leading cause of death in the United States. It has been well established that an adverse intrauterine environment increases vulnerability to cardiovascular disease later in life [1, 2]. Environmental factors during the critical period of fetal development can influence the maturation of organs, such as the heart. Involved in this maturation is a transition of cardiomyocytes from a mononucleate to a binucleate phenotype. This normal transition occurs during fetal and early postnatal life, and is attributed to the uncoupling of cytokinesis from karyokinesis [3]. Mononucleate cardiomyocytes retain the ability to proliferate whereas the binucleate cells do not, and this is because they have exited the cell cycle and become terminally differentiated [4]. Alterations to the timing of this critical transition may have long-term consequences on heart development and function throughout life.
Hypoxia is a major stress to the fetal development. Our previous studies have shown that an in vivo rat model of maternal hypoxia results in fetal cardiomyocytes prematurely exiting the cell cycle [5-7]. This early-onset transition leads to fewer but larger cardiomyocytes as a result of increased binucleation and hypertrophy, and decreased proliferation of the cells. The timing of this transition is critical in determining the number of cardiomyocytes endowed in the heart for a lifetime. Whereas these in vivo studies showed the effect of hypoxia on fetal heart development, the mechanisms remain unknown.
Hypoxia is a known inducer of endothelin-1 (ET-1) expression [8-11]. ET-1 plays an important role in regulating cell cycle, and the cardiomyocyte is both a site of synthesis and action of ET-1 [12, 13], suggesting a localized role for hypoxia-induced ET-1 action in the heart. Thus, the present study tested the hypothesis that ET-1 induces a premature cardiomyocyte transition in the developing heart. Given a recent finding that the terminal differentiation of cardiomyocytes is characterized by a hypermethylated genome and compact chromatin [14], we further tested the hypothesis that ET-1 promotes cardiomyocyte terminal differentiation by an increase in DNA methylation. Herein, we present evidence that ET-1 via action of ET-1 receptors stimulates the premature transition of fetal cardiomyocytes, characterized by increased binucleation and decreased proliferation. DNA methylation of fetal cardiomyocytes is increased with ET-1 treatment, and the ET-1-induced changes in binucleation and proliferation are blocked by a DNA methylation inhibitor 5-aza-2'-deoxycytidine. Altogether the results suggest that epigenetic regulation via DNA methylation is involved in the cardiomyocyte transition stimulated by increased synthesis of ET-1.
Methods
Experimental animals
Time-dated pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Portage, MI) and divided into two groups: (i) normoxic control and (ii) 10.5% O2 hypoxia treatment from gestational day 15 to 21, as previously described [15, 16]. Hearts were isolated from day 21 fetuses. To isolate hearts, pregnant rats were anesthetized with isoflurane, and adequate anesthesia was determined by loss of pedal withdrawal reflex. Fetuses were removed and pregnant rats killed by removing the hearts. Fetal hearts were isolated for the studies. All procedures and protocols were approved by the Institutional Animal Care and Use Committee and followed the guidelines by US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Primary cardiomyocyte culture and treatment
Cardiomyocytes were isolated from normoxic day 21 fetal rats as previously described [17]. Cells were cultured in Hyclone Medium 199 (Thermo Scientific) supplemented with 10% fetal bovine serum (Gemini Bio-Products) and 1% antibiotics (10,000 I.U./mL penicillin, 10,000 μg/mL streptomycin) at 37ºC in 95% air/5% CO2. BrdU (0.1mM) was added to the medium to prevent fibroblast proliferation. Within three days of culture, the cells formed a monolayer with synchronized beating, characteristic of viable cardiomyocytes. Experiments were performed at 70-80% confluency. Cells were treated under normoxia (21% O2) or hypoxia (1% O2) for 24 hours, in the absence or presence of ET-1 (Sigma; 10 nM), PD145065 (Calbiochem; 10 nM), or 5-aza-2'-deoxycytidine (Sigma; 10 μM).
Quantitative real-time PCR
RNA was isolated from the fetal hearts and prepro-ET-1 mRNA abundance was determined by real-time RT-PCR using Icycler Thermal cycler (Bio-Rad), as described previously [16]. Reverse transcription and cDNA synthesis was performed using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). The primers are 5'-CTAGGTCTAAGCGATCCTTGAA-3' (forward) and 5'-CTTGATGCTGTTGCTGATGG-3' (reverse). PCR was performed in triplicate, and threshold cycle numbers were averaged.
Immunocytochemistry
Primary cardiomyocytes were double stained with alpha-actinin, a cardiomyocyte marker, and Ki-67, a proliferation marker as described previously [5]. Cardiomyocytes were plated on coverslips and fixed with acetone for 10 minutes. The cells were blocked with 1% bovine serum albumin for 1 hour at room temperature before incubation with the primary antibodies: mouse anti-α-sarcomeric actinin (Sigma, St.Louis, MO) (1:200) and rabbit anti-Ki-67 (Abcam, Cambridge, MA) (1:100) in 4ºC overnight. The samples were incubated with the secondary antibodies: anti-mouse FITC-conjugated and anti-rabbit Texas Red-conjugated antibodies for 1 hour at room temperature. Nuclei were stained with Hoescht (Sigma) for 1 minute. The immunofluorescence staining was assessed using a Zeiss Axio Imager.A1 microscope and quantitative analysis was carried out using Image J software. Percent binucleation, Ki-67 expression, and cell size were measured.
5-mC DNA ELISA
DNA methylation in primary fetal cardiomyocytes was determined by measuring 5-methylcytosine (5-mC) using a 5-mC DNA ELISA kit (Zymo Research). The kit features a unique anti-5-mC monoclonal antibody that is both sensitive and specific for 5-mC. The protocol for measurement of 5-mC level is described in the manufacturer's instruction. Briefly, 100 ng of genomic DNA from cardiomyocytes and standard controls provided by the kit was denatured and used to coat the plate wells with 5-mC coating buffer. After incubation at 37°C for 1 hour, the wells were washed with 5-mC ELISA buffer and then an antibody mix consisting of anti-5-mC and a secondary antibody was added to each well. The plate was covered with foil and incubated at 37°C for 1 hour. After the antibody mix was washed out from the wells with the 5-mC ELISA buffer, a HRP developer was added to each well and incubated at room temperature for 1 hour. The absorbance at 405 nm was measured using an ELISA plate reader. The percent 5-mC was calculated using the second-order regression equation of the standard curve that was constructed with negative control and positive controls in the same experiment.
Statistical Analysis
Data are expressed as means ± SEM. Statistical analysis (p < 0.05) was determined by analysis of variance followed by Neuman-Keuls post hoc test or Student's t test, where appropriate.
Results
Maternal hypoxia increased prepro-ET-1 mRNA in fetal hearts
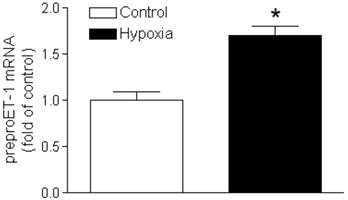
Animals were exposed to maternal hypoxia from gestational day 15-21; at the end of treatment hearts were isolated from day 21 fetal rats. Figure 1 demonstrated a significant increase in prepro-ET-1 mRNA abundance in fetal hearts exposed to 10.5% O2, as compared to the normoxic control (21% O2).
Effect of hypoxia on prepro-ET1 mRNA in the fetal heart. Hearts were isolated from near-term fetuses of pregnant rats treated with control or hypoxia. mRNA abundance of prepro-ET-1 was determined by real-time RT-PCR. Data are means ± SEM. * P < 0.05, hypoxia vs. control. n = 7-8
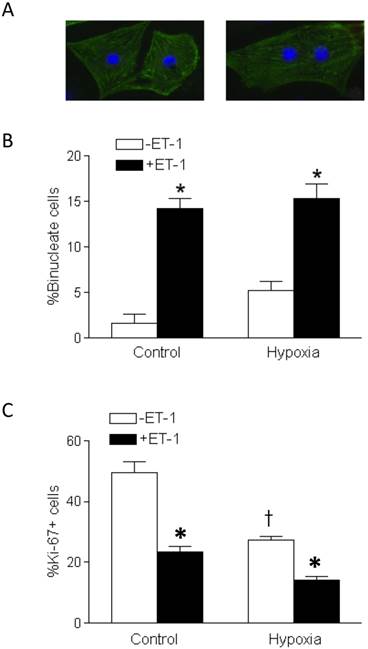
ET-1 stimulated binucleation and inhibited proliferation of fetal cardiomyocytes
The morphology of mononucleate cells and binucleate cells of primary fetal cardiomyocytes are shown in Figure 2A. Whereas the basal levels of binucleate cardiomyocytes in fetal hearts were low, the treatment of cardiomyocytes with ET-1 resulted in a significant increase in percent binucleation, as compared to the control in both normoxic and hypoxic conditions (Figure 2A). Hypoxia alone in the absence of ET-1 had a slight increase in percent binucleation but it did not reach a significant level, as compared to the normoxic control. Percent Ki-67 positive cells, indicating proliferation, in ET-1 treated cardiomyocytes were significantly decreased in both normoxia and hypoxia (Figure 2B). Unlike the effect of binucleation, hypoxia alone in the absence of ET-1 significantly decreased percent of Ki-67 positive cells (Figure 2B).
Effect of ET-1 on binucleation and proliferation of fetal cardiomyocytes. Cardiomyocytes isolated from fetal hearts were treated with ET-1 (10 nM) under normoxic control (21% O2) or hypoxic (1% O2) conditions for 24 h. A. Morphology of mononucleate and binucleate fetal cardiomyocytes. B. Binucleation result. C. Proliferation result. Data are means ± SEM. * P < 0.05, +ET-1 vs. -ET-1; † P < 0.05, hypoxia vs. control. n = 5
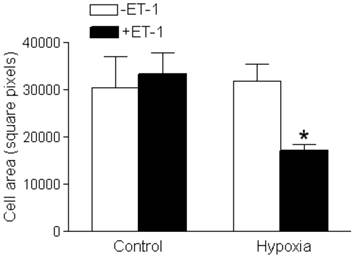
Interaction of ET-1 and hypoxia decreased cardiomyocyte size
Neither ET-1 nor hypoxia alone had a significant effect on cardiomyocyte size (Figure 3). However, cardiomyocyte size was significantly decreased with the ET-1 treatment under the hypoxic condition (Figure 3).
Effect of ET-1 on fetal cardiomyocyte size. Cardiomyocytes isolated from fetal hearts were treated with ET-1 (10 nM) under normoxic control (21% O2) or hypoxic (1% O2) conditions for 24 h. Data are means ± SEM. * P < 0.05, +ET-1 vs. -ET-1. n = 7-10
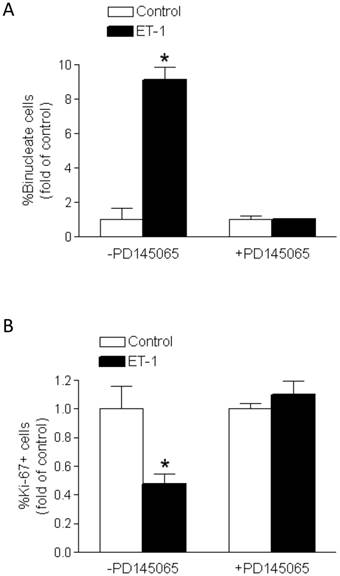
PD145065 inhibited the effects of ET-1
PD145065, a non-selective ET-receptor antagonist, blocked the effects of ET-1 on percent binucleation (Figure 4A) and Ki-67 expression (Figure 4B) in fetal cardiomyocytes. PD145065 in the absence of ET-1 had no significant effect on either binucleation or proliferation of cardiomyocytes.
PD145065 abrogates ET-1-mediated effects on binucleation and proliferation of fetal cardiomyocytes. Cardiomyocytes isolated from fetal hearts were treated with ET-1 (10 nM) for 24 h in the absence or presence of PD145065 (10 nM). A. Binucleation result. B. Proliferation result. * P < 0.05, ET-1 vs. control. n = 5
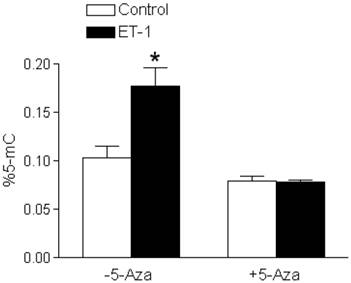
ET-1 increased DNA methylation in fetal cardiomyocytes
ET-1 treatment of fetal cardiomyocytes resulted in a significant increase in DNA methylation, seen as increased percent 5-mC in Figure 5. In the presence of 5-aza-2'-deoxycytidine, a DNA methylation inhibitor, the effects of ET-1 were blocked (Figure 5).
5-Aza-2'-deoxycytidine blocks ET-1-increased DNA methylation in fetal cardiomyocytes. Cardiomyocytes isolated from fetal hearts were treated with ET-1 (10 nM) for 24 h in the absence or presence of 5-aza-2'-deoxycytidine (5-Aza, 10 µM). * P < 0.05, ET-1 vs. control. n = 5
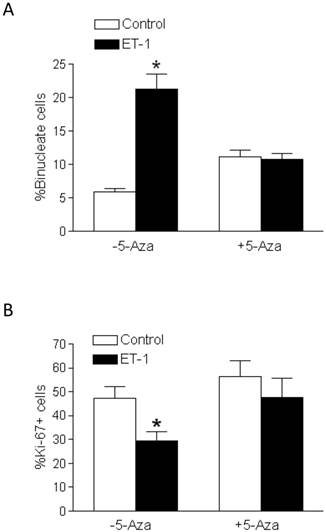
5-Aza-2'-deoxycytidine abrogated the effects of ET-1 on binucleation and proliferation in fetal cardiomyocytes
In the presence of 5-aza-2'-deoxycytidine, ET-1-induced stimulation of binucleation (Figure 6A) and inhibition of Ki-67 expression (Figure 6B) in fetal cardiomyocytes were blocked. Whereas 5-aza-2'-deoxycytidine alone in the absence of ET-1 had a tendency to increase cardiomyocyte binucleation, this effect did not reach the significant level (Figure 6A).
5-Aza-2'-deoxycytidine abrogates ET-1-mediated effects on binucleation and proliferation of fetal cardiomyocytes. Cardiomyocytes isolated from fetal hearts were treated with ET-1 (10 nM) for 24 h in the absence or presence of 5-aza-2'-deoxycytidine (5-Aza, 10 µM). A. Binucleation result. B. Proliferation result. * P < 0.05, ET-1 vs. control. n = 5
Discussion
The present study provides evidence that ET-1 inhibited proliferation and induced the premature transition of fetal cardiomyocytes from a mononucleate to a binucleate phenotype, indicative of terminally differentiated cardiomyocytes. Hypoxia alone did not elicit the same effects as ET-1. An ET-receptor antagonist, PD145065 blocked the ET-1-induced increase in binucleation and decrease in proliferation. Additionally, we demonstrated that ET-1 treatment increased DNA methylation in fetal cardiomyocytes, and a DNA methylation inhibitor, 5-aza-2'-deoxycytidine abrogated ET-1-induced DNA methylation and terminal differentiation of cardiomyocytes.
The ET-1 synthesis pathway begins with the transcription of prepro-ET-1 mRNA, which is translated into prepro-ET-1. A series of enzymatic cleavages produce Big-ET-1 and the matured ET-1, a 21-amino acid peptide [18]. Several studies have reported the regulation of ET-1 synthesis by hypoxia, including the identification of a HIF-1 binding site on the prepro-ET-1 gene promoter [19, 20]. Specifically in cardiomyocytes, a HIF-1α binding site has been identified on the 5'-promoter region of the prepro-ET-1 gene [21]. Additionally, previous studies demonstrated a negative correlation between pO2 and plasma ET-1 levels in fetal goats [11] as well as pO2 and ET-1 levels in human amniotic fluid [10]. ET-1 mRNA levels were significantly increased in rat lung [22] and placentas [23] exposed to hypoxia. In agreement with these findings, the present study demonstrated a significant increase in prepro-ET-1 mRNA in the fetal rat heart resulting from in utero hypoxia, suggesting a local paracrine action of ET-1 in hypoxia-mediated effect on the fetal heart.
Physiological circulating levels of ET-1 are in the low picomolar range [12, 24] and may be significantly upregulated in pathophysiological conditions, such as hypoxia, heart failure, hypertension, and preeclampsia [25-28]. ET-1 acts in a paracrine and/or autocrine fashion and therefore tissue concentrations are significantly higher than those in the circulation [29]. The concentration of ET-1 (10 nM) was chosen based on other studies [30-33] and the rationale that ET-1 levels in the local tissue are much greater than in circulation. In the present study, we found that fetal rat cardiomyocytes exposed to elevated levels of ET-1 exhibited both increased binucleation and decreased proliferation. The binucleate cells are unable to proliferate and thus become terminally differentiated. In the rat heart, normal transition of cardiomyocytes to the binucleate form starts at birth and lasts during the first two weeks of postnatal life [3]. Therefore, the accelerated transition in the fetal heart due to increased levels of ET-1 has long-term implications. A premature transition of terminal differentiation may result in a reduced number of cardiomyocytes and altered cardiac growth after birth. As previous work has shown, hypoxia causes a premature exit of cell cycle in fetal cardiomyocytes [7], but the downstream regulators are not known.
The present study demonstrated that ET-1 increased the percent of binucleate cells independent of hypoxia, and hypoxia alone had no significant effect on the binucleation of cardiomyocytes. These findings suggest a lack of direct effect of hypoxia on the cardiomyocyte transition and provide evidence that ET-1 is a key downstream regulator of the premature exit of cell cycle in the fetal heart, observed in vivo in fetal hypoxia [7]. It is important to note that while cardiomyocytes have the ability to produce ET-1, endothelial cells contribute a large portion of its production. The isolated cardiomyocytes may not provide the full mechanism for hypoxia to produce a sufficient ET-1 response and the subsequent effects on binucleation and cell size. The finding that PD145065 blocked the ET-1-induced increase in binucleation and decrease in proliferation of fetal cardiomyocytes indicates the ET-1 receptor-mediated effects. PD145065 is a non-selective ETA- and ETB-receptor antagonist and has been shown to block the effect of ET-1 via the ET-receptors [34-36].
The finding that hypoxia decreased proliferation of fetal cardiomyocytes is in agreement with previous reports [5-7]. Prior studies have shown that hypoxia promotes HIF-1α association with HIF-1β and enhances the expression of cyclin-dependent kinase inhibitors (CKIs), which in turn inhibits cell cycle proteins and decreases cell proliferation [37]. It is also suggested that HIF-1 regulates the G1/S phase transition by regulating the expression of cyclin E, a required factor for the transition [38]. Hypoxia has also been shown to induce expression of metalloproteinase inhibitors (TIMPs) that may have an inhibitory or stimulatory effect on cellular proliferation depending on the subtype and tissue involved [39]. In the heart, TIMP-3 is highly expressed and shown to inhibit proliferation in neonatal mouse cardiomyocytes [40]; a result of up-regulated p27 expression via the EGFR-JNK-SP-1 mediated pathway [41]. Although TIMP-3 and -4 are upregulated by hypoxia, their promoters do not contain HIF-responsive elements. Thus the regulation of these inhibitors and the subsequent effects on proliferation appear to be mediated by other genes that contain HIF-responsive elements.
ET-1 is one possible candidate considering it both contains a HIF-response element in its promoter [19, 20] and has been shown to regulate proliferation [5-7, 42]. The finding that, unlike ET-1, hypoxia had no significant effect on binucleation suggests that hypoxia-induced effect on proliferation was not mediated by ET-1, but rather by an independent and direct effect of hypoxia. The synergistic effect of hypoxia and ET-1 more closely mimics the physiological system as a whole. Ki-67 expression and cell size were significantly decreased by hypoxia and ET-1 treatment together. These results agree with previous reports, from our lab and others, that hypoxia alone had a direct effect in decreasing proliferation of cardiomyocytes [5-7, 42].
A change in cell size was only observed with the addition of both ET-1 and hypoxia. Both ET-1 and hypoxia are known hypertrophic factors [43-45]. In neonatal rat cardiomyocytes, mild hypoxia (10% O2) has been found to induce hypertrophy [45, 46]. However more severe hypoxia (1% O2), as was done in our study, appears to elicit the opposite effect leading to a reduction in cell size. Thus the severity at which hypoxia is induced likely has a differential effect on changes in cellular size. ET-1 has also been shown to stimulate cardiomyocytes to proliferate, and in the case of terminally differentiated cells it leads to hypertrophy [32, 47]. Furthermore previous studies have found that hypertrophic growth is initially observed in the first week of postnatal life [3, 48]. Given that fetal and neonatal hearts are at very different developmental stages and experience very different oxygen tensions, it is possible that cardiomyocytes of the fetal heart respond differently to environmental cues such as hypoxia as that seen in the neonatal cardiomyocytes.
The finding that 5-aza-2'-deoxycytidine blocked the ET-1-induced increase in binucleation and decrease in proliferation is intriguing and suggests that ET-1 induces methylation of DNA as a means of involvement in cardiomyocyte terminal differentiation and suppression of proliferation. Whereas the present study focused on the downstream mechanisms of ET-1 in regulating terminal differentiation of cardiomyocytes, whether DNA methylation plays a role in the hypoxia-mediated direct effect on proliferation remains to be determined. 5-Aza-2'-deoxycytidine has been widely used as a DNA methylation inhibitor, and in the concentration range of 1 to 30 µM it inhibits DNA methylation both globally and at specific sites of DNA [14, 49-52]. In the present study, we found that ET-1 significantly increased global DNA methylation in cardiomyocytes and this was blocked by 5-aza-2'-deoxycytidine. Epigenetic mechanism of DNA methylation acts to silence gene transcription, typically at cytosine residues within CpG dinucleotides. A previous study showed that methylation gradually increases over the course of development in neonatal cardiomyocytes [14], the same time frame for which binucleation occurs. Furthermore, expression of DNA methyltransferases involved in de novo DNA methylation (DNMT3a and DNMT3b) was significantly increased during the first 90 days of postnatal life. Inhibition of methylation with 5-aza-2'-deoxycytidine during neonatal day 7 and 10 resulted in a marked increase in DNA synthesis and delayed maturation [14]. It is well known that environmental cues during fetal development can profoundly alter the structure and function of an organ via epigenetic regulation. Particularly in the heart, cardiac function is dependent in part on cardiomyocyte number. Thus hypoxia-mediated ET-1 may signal through epigenetic mechanisms to negatively impact cardiomyocyte development. The present study suggests that DNA methylation is an epigenetic mechanism through which ET-1 stimulates cardiomyocyte transition of terminal differentiation. Ultimately, this may lead to reduced total cardiomyocyte number in the heart. Many studies have demonstrated that genes associated with the cell cycle and cytokinesis are involved in this transition process [53-57]. Adult cardiomyocytes from knockout mice lacking Rb and p130 show a decrease in heterochromatin and an increase in proliferation associated with derepression of cell cycle genes [57]. These genes may be differentially regulated by changes in methylation patterns thus altering the cell cycle and cytokinesis. Future studies will have to elucidate the methylation status of specific genes during this transition phase.
The present study identifies a novel mechanism of ET-1-induced hypermethylation as a downstream regulator of hypoxia-mediated cardiomyocyte transition from mononucleate to binucleate cells in the developing heart. Cardiomyocyte endowment is determined during fetal and early postnatal development, when most cardiomyocytes become binucleate and cease to proliferate [58, 59]. Given that hypoxia is one of the most important and clinically relevant stresses to the fetal development, and that fetal hypoxia results in fewer but larger cardiomyocytes and increases the susceptibility of the heart to ischemic injury in offspring [7, 60-62], the present study provides a mechanistic understanding worthy of further investigation in humans.
Acknowledgements
A portion of this research used the Loma Linda University School of Medicine Advanced Imaging and Microscopy Core, a facility supported in part by the National Science Foundation through the Major Research Instrumentation program of the Division of Biological Infrastructure Grant No. 0923559 and the Loma Linda University School of Medicine.
Funding
This study was supported by the National Institutes of Health grants HL82779 (L.Z.), HL83966 (L.Z.), DA032510 (D.X.), and California Tobacco-Related Disease Research Program Award 22XT-0022 (D.X.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Barker DJ. Fetal origins of coronary heart disease. BMJ (Clinical research ed). 1995;311:171-4
2. Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077-81
3. Clubb FJJr, Bishop SP. Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Laboratory investigation, a journal of technical methods and pathology. 1984;50:571-7
4. Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiological reviews. 2007;87:521-44 doi:10.1152/physrev.00032.2006
5. Tong W, Xiong F, Li Y, Zhang L. Hypoxia inhibits cardiomyocyte proliferation in fetal rat hearts via upregulating TIMP-4. American journal of physiology Regulatory, integrative and comparative physiology. 2013 doi:10.1152/ajpregu.00515.2012
6. Tong W, Xue Q, Li Y, Zhang L. Maternal hypoxia alters matrix metalloproteinase expression patterns and causes cardiac remodeling in fetal and neonatal rats. American journal of physiology Heart and circulatory physiology. 2011;301:H2113-21 doi:10.1152/ajpheart.00356.2011
7. Bae S, Xiao Y, Li G, Casiano CA, Zhang L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. American journal of physiology Heart and circulatory physiology. 2003;285:H983-90 doi:10.1152/ajpheart.00005.2003
8. Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. The Journal of biological chemistry. 2001;276:12645-53 doi:10.1074/jbc.M011344200
9. Hashiguchi K, Takagi K, Nakabayashi M, Takeda Y, Sakamoto S, Naruse M. et al. Relationship between fetal hypoxia and endothelin-1 in fetal circulation. Journal of cardiovascular pharmacology. 1991;17(Suppl 7):S509-10
10. Ostlund E, Lindholm H, Hemsen A, Fried G. Fetal erythropoietin and endothelin-1: relation to hypoxia and intrauterine growth retardation. Acta obstetricia et gynecologica Scandinavica. 2000;79:276-82
11. Yamada J, Fujimori K, Ishida T, Sanpei M, Honda S, Sato A. Plasma endothelin-1 and atrial natriuretic peptide levels during prolonged (24-h) non-acidemic hypoxemia in fetal goats. The Journal of maternal-fetal medicine. 2001;10:409-13
12. Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiological reviews. 2011;91:1-77 doi:10.1152/physrev.00060.2009
13. Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annual review of pharmacology and toxicology. 2001;41:851-76 doi:10.1146/annurev.pharmtox.41.1.851
14. Kou CY, Lau SL, Au KW, Leung PY, Chim SS, Fung KP. et al. Epigenetic regulation of neonatal cardiomyocytes differentiation. Biochemical and biophysical research communications. 2010;400:278-83 doi:10.1016/j.bbrc.2010.08.064
15. Patterson AJ, Xiao D, Xiong F, Dixon B, Zhang L. Hypoxia-derived oxidative stress mediates epigenetic repression of PKCepsilon gene in foetal rat hearts. Cardiovascular research. 2012;93:302-10 doi:10.1093/cvr/cvr322
16. Xue Q, Dasgupta C, Chen M, Zhang L. Foetal hypoxia increases cardiac AT(2)R expression and subsequent vulnerability to adult ischaemic injury. Cardiovascular research. 2011;89:300-8 doi:10.1093/cvr/cvq303
17. Xiao Y, He J, Gilbert RD, Zhang L. Cocaine induces apoptosis in fetal myocardial cells through a mitochondria-dependent pathway. The Journal of pharmacology and experimental therapeutics. 2000;292:8-14
18. Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Canadian journal of physiology and pharmacology. 2008;86:485-98 doi:10.1139/y08-059
19. Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochemical and biophysical research communications. 1998;245:894-9 doi:10.1006/bbrc.1998.8543
20. Minchenko A, Caro J. Regulation of endothelin-1 gene expression in human microvascular endothelial cells by hypoxia and cobalt: role of hypoxia responsive element. Molecular and cellular biochemistry. 2000;208:53-62
21. Kakinuma Y, Miyauchi T, Yuki K, Murakoshi N, Goto K, Yamaguchi I. Novel molecular mechanism of increased myocardial endothelin-1 expression in the failing heart involving the transcriptional factor hypoxia-inducible factor-1alpha induced for impaired myocardial energy metabolism. Circulation. 2001;103:2387-94
22. Li H, Chen SJ, Chen YF, Meng QC, Durand J, Oparil S. et al. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. Journal of applied physiology (Bethesda, Md: 1985). 1994;77:1451-9
23. Thaete LG, Jilling T, Synowiec S, Khan S, Neerhof MG. Expression of endothelin 1 and its receptors in the hypoxic pregnant rat. Biology of reproduction. 2007;77:526-32 doi:10.1095/biolreprod.107.061820
24. Nakas-Icindic E, Zaciragic A, Hadzovic A, Avdagic N. Endothelin in health and disease. Bosnian journal of basic medical sciences / Udruzenje basicnih mediciniskih znanosti = Association of Basic Medical Sciences. 2004;4:31-4
25. George EM, Granger JP. Endothelin: key mediator of hypertension in preeclampsia. American journal of hypertension. 2011;24:964-9 doi:10.1038/ajh.2011.99
26. Rautureau Y, Schiffrin EL. Endothelin in hypertension: an update. Current opinion in nephrology and hypertension. 2012;21:128-36 doi:10.1097/MNH.0b013e32834f0092
27. Wei CM, Lerman A, Rodeheffer RJ, McGregor CG, Brandt RR, Wright S. et al. Endothelin in human congestive heart failure. Circulation. 1994;89:1580-6
28. Zolk O, Quattek J, Sitzler G, Schrader T, Nickenig G, Schnabel P. et al. Expression of endothelin-1, endothelin-converting enzyme, and endothelin receptors in chronic heart failure. Circulation. 1999;99:2118-23
29. Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacological reviews. 1994;46:325-415
30. Li X, Wei XL, Meng LL, Chi MG, Yan JQ, Ma XY. et al. Involvement of tissue transglutaminase in endothelin 1-induced hypertrophy in cultured neonatal rat cardiomyocytes. Hypertension. 2009;54:839-44 doi:10.1161/hypertensionaha.109. 130161
31. Majumdar P, Chen S, George B, Sen S, Karmazyn M, Chakrabarti S. Leptin and endothelin-1 mediated increased extracellular matrix protein production and cardiomyocyte hypertrophy in diabetic heart disease. Diabetes/metabolism research and reviews. 2009;25:452-63 doi:10.1002/dmrr.964
32. Ito H, Hirata Y, Hiroe M, Tsujino M, Adachi S, Takamoto T. et al. Endothelin-1 induces hypertrophy with enhanced expression of muscle-specific genes in cultured neonatal rat cardiomyocytes. Circulation research. 1991;69:209-15
33. Yu L, Li M, She T, Shi C, Meng W, Wang B. et al. Endothelin-1 Stimulates the Expression of L-Type Ca(2+) Channels in Neonatal Rat Cardiomyocytes via the Extracellular Signal-Regulated Kinase 1/2 Pathway. The Journal of membrane biology. 2013;246:343-53 doi:10.1007/s00232-013-9538-7
34. Ceccarelli F, Scavuzzo MC, Giusti L, Bigini G, Costa B, Carnicelli V. et al. ETA receptor-mediated Ca2+ mobilisation in H9c2 cardiac cells. Biochemical pharmacology. 2003;65:783-93
35. Drimal J, Knezl V, Drimal J Jr, Drimal D, Bauerova K, Kettmann V. et al. Cardiac effects of endothelin-1 (ET-1) and related C terminal peptide fragment: increased inotropy or contribution to heart failure? Physiological research / Academia Scientiarum Bohemoslovaca. 2003;52:701-8
36. Doherty AM, Cody WL, He JX, DePue PL, Cheng XM, Welch KM. et al. In vitro and in vivo studies with a series of hexapeptide endothelin antagonists. Journal of cardiovascular pharmacology. 1993;22(Suppl 8):S98-102
37. Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Molecular and cellular biology. 2003;23:359-69
38. Goda N, Dozier SJ, Johnson RS. HIF-1 in cell cycle regulation, apoptosis, and tumor progression. Antioxidants & redox signaling. 2003;5:467-73 doi:10.1089/152308603768295212
39. Tong W, Zhang L. Fetal hypoxia and programming of matrix metalloproteinases. Drug discovery today. 2012;17:124-34 doi:10.1016/j.drudis.2011.09.011
40. Hammoud L, Xiang F, Lu X, Brunner F, Leco K, Feng Q. Endothelial nitric oxide synthase promotes neonatal cardiomyocyte proliferation by inhibiting tissue inhibitor of metalloproteinase-3 expression. Cardiovascular research. 2007;75:359-68 doi:10.1016/j.cardiores.2007.05.006
41. Hammoud L, Burger DE, Lu X, Feng Q. Tissue inhibitor of metalloproteinase-3 inhibits neonatal mouse cardiomyocyte proliferation via EGFR/JNK/SP-1 signaling. American journal of physiology Cell physiology. 2009;296:C735-45 doi:10.1152/ajpcell.00246.2008
42. Zhang FX, Chen ML, Shan QJ, Zou JG, Chen C, Yang B. et al. Hypoxia reoxygenation induces premature senescence in neonatal SD rat cardiomyocytes. Acta pharmacologica Sinica. 2007;28:44-51 doi:10.1111/j.1745-7254.2007.00488.x
43. Suzuki T, Hoshi H, Mitsui Y. Endothelin stimulates hypertrophy and contractility of neonatal rat cardiac myocytes in a serum-free medium. FEBS letters. 1990;268:149-51
44. Shubeita HE, McDonough PM, Harris AN, Knowlton KU, Glembotski CC, Brown JH. et al. Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular myocytes. A paracrine mechanism for myocardial cell hypertrophy. The Journal of biological chemistry. 1990;265:20555-62
45. Ito H, Adachi S, Tamamori M, Fujisaki H, Tanaka M, Lin M. et al. Mild hypoxia induces hypertrophy of cultured neonatal rat cardiomyocytes: a possible endogenous endothelin-1-mediated mechanism. Journal of molecular and cellular cardiology. 1996;28:1271-7 doi:10.1006/jmcc.1996.0117
46. Chu W, Wan L, Zhao D, Qu X, Cai F, Huo R. et al. Mild hypoxia-induced cardiomyocyte hypertrophy via up-regulation of HIF-1alpha-mediated TRPC signalling. Journal of cellular and molecular medicine. 2012;16:2022-34 doi:10.1111/j.1582-4934.2011.01497.x
47. Cullingford TE, Markou T, Fuller SJ, Giraldo A, Pikkarainen S, Zoumpoulidou G. et al. Temporal regulation of expression of immediate early and second phase transcripts by endothelin-1 in cardiomyocytes. Genome biology. 2008;9:R32. doi:10.1186/gb-2008-9-2-r32
48. Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. Journal of molecular and cellular cardiology. 1996;28:1737-46 doi:10.1006/jmcc.1996.0163
49. Meyer K, Zhang H, Zhang L. Direct effect of cocaine on epigenetic regulation of PKCepsilon gene repression in the fetal rat heart. Journal of molecular and cellular cardiology. 2009;47:504-11 doi:10.1016/j.yjmcc.2009.06.004
50. Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKC{epsilon} gene repression in rat hearts. Circulation research. 2010;107:365-73 doi:10.1161/circresaha.110.221259
51. Vallender TW, Lahn BT. Localized methylation in the key regulator gene endothelin-1 is associated with cell type-specific transcriptional silencing. FEBS letters. 2006;580:4560-6 doi:10.1016/j.febslet.2006.07.017
52. Xiong F, Xiao D, Zhang L. Norepinephrine causes epigenetic repression of PKCε gene in rodent hearts by activating Nox1-dependent reactive oxygen species production. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26:2753-63 doi:10.1096/fj.11-199422
53. Liu Y, Tang MK, Cai DQ, Li M, Wong WM, Chow PH. et al. Cyclin I and p53 are differentially expressed during the terminal differentiation of the postnatal mouse heart. Proteomics. 2007;7:23-32 doi:10.1002/pmic.200600456
54. Ahuja P, Perriard E, Pedrazzini T, Satoh S, Perriard JC, Ehler E. Re-expression of proteins involved in cytokinesis during cardiac hypertrophy. Experimental cell research. 2007;313:1270-83 doi:10.1016/j.yexcr.2007.01.009
55. Chen HW, Yu SL, Chen WJ, Yang PC, Chien CT, Chou HY. et al. Dynamic changes of gene expression profiles during postnatal development of the heart in mice. Heart (British Cardiac Society). 2004;90:927-34 doi:10.1136/hrt.2002.006734
56. Engel FB, Schebesta M, Keating MT. Anillin localization defect in cardiomyocyte binucleation. Journal of molecular and cellular cardiology. 2006;41:601-12 doi:10.1016/j.yjmcc.2006.06.012
57. Sdek P, Zhao P, Wang Y, Huang CJ, Ko CY, Butler PC. et al. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. The Journal of cell biology. 2011;194:407-23 doi:10.1083/jcb.201012049
58. Botting KJ, Wang KC, Padhee M, McMillen IC, Summers-Pearce B, Rattanatray L. et al. Early origins of heart disease: low birth weight and determinants of cardiomyocyte endowment. Clinical and experimental pharmacology & physiology. 2012;39:814-23 doi:10.1111/j.1440-1681.2011.05649.x
59. Thornburg K, Jonker S, O'Tierney P, Chattergoon N, Louey S, Faber J. et al. Regulation of the cardiomyocyte population in the developing heart. Progress in biophysics and molecular biology. 2011;106:289-99 doi:10.1016/j.pbiomolbio.2010.11.010
60. Li G, Bae S, Zhang L. Effect of prenatal hypoxia on heat stress-mediated cardioprotection in adult rat heart. American journal of physiology Heart and circulatory physiology. 2004;286:H1712-9 doi:10.1152/ajpheart.00898.2003
61. Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. Journal of the Society for Gynecologic Investigation. 2003;10:265-74
62. Xu Y, Williams SJ, O'Brien D, Davidge ST. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:1251-3 doi:10.1096/fj.05-4917fje
Author contact
![]() Corresponding author: Lubo Zhang, Ph.D., Center for Perinatal Biology, Division of Pharmacology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350. Tel: 909-558-4325; Fax: 909-558-4029; email: lzhangedu
Corresponding author: Lubo Zhang, Ph.D., Center for Perinatal Biology, Division of Pharmacology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350. Tel: 909-558-4325; Fax: 909-558-4029; email: lzhangedu