Impact Factor ISSN: 1449-1907
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(12):1727-1739. doi:10.7150/ijms.6884 This issue Cite
Research Paper
DNA Methylation Profiling Revealed Promoter Hypermethylation-induced Silencing of p16, DDAH2 and DUSP1 in Primary Oral Squamous Cell Carcinoma
Goot Heah Khor1,2,3,8 ![]() , Gabriele Ruth Anisah Froemming2, Rosnah Binti Zain3,4, Mannil Thomas Abraham5, Effat Omar2, Su Keng Tan6, Aik Choon Tan7, Vui King Vincent-Chong3, Kwai Lin Thong8,9
, Gabriele Ruth Anisah Froemming2, Rosnah Binti Zain3,4, Mannil Thomas Abraham5, Effat Omar2, Su Keng Tan6, Aik Choon Tan7, Vui King Vincent-Chong3, Kwai Lin Thong8,9 ![]()
1. Centre of Studies for Preclinical Science, Faculty of Dentistry, Universiti Teknologi MARA, Shah Alam. Selangor, Malaysia.
2. Institute of Medical Molecular Biotechnology, Faculty of Medicine, Universiti Teknologi MARA, Selangor, Malaysia.
3. Oral Cancer Research and Coordinating Center, Faculty of Dentistry, University of Malaya, Kuala Lumpur, Malaysia.
4. Department of Oro-maxillofacial Surgical and Medical Sciences, Faculty of Dentistry, University of Malaya, Kuala Lumpur, Malaysia.
5. Tengku Ampuan Rahimah Hospital, Department of Oral and Maxillofacial Surgery, Ministry of Health Malaysia, Malaysia.
6. Oral Maxillofacial Diagnostic and Medicine, Faculty of Dentistry, Universiti Teknologi MARA, Shah Alam. Selangor, Malaysia.
7. Division of Medical Oncology, Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA.
8. Laboratory of Biomedical Science and Molecular Microbiology, UMBIO Cluster, Institute of Postgraduate, University of Malaya, Kuala Lumpur, Malaysia.
9. Institute of Biological Science, Faculty of Science, University of Malaya, Kuala Lumpur, Malaysia.
Received 2013-6-11; Accepted 2013-9-9; Published 2013-10-12
Abstract
Background: Hypermethylation in promoter regions of genes might lead to altered gene functions and result in malignant cellular transformation. Thus, biomarker identification for hypermethylated genes would be very useful for early diagnosis, prognosis, and therapeutic treatment of oral squamous cell carcinoma (OSCC). The objectives of this study were to screen and validate differentially hypermethylated genes in OSCC and correlate the hypermethylation-induced genes with demographic, clinocopathological characteristics and survival rate of OSCC.
Methods: DNA methylation profiling was utilized to screen the differentially hypermethylated genes in OSCC. Three selected differentially-hypermethylated genes of p16, DDAH2 and DUSP1 were further validated for methylation status and protein expression. The correlation between demographic, clinicopathological characteristics, and survival rate of OSCC patients with hypermethylation of p16, DDAH2 and DUSP1 genes were analysed in the study.
Results: Methylation profiling demonstrated 33 promoter hypermethylated genes in OSCC. The differentially-hypermethylated genes of p16, DDAH2 and DUSP1 revealed positivity of 78%, 80% and 88% in methylation-specific polymerase chain reaction and 24% and 22% of immunoreactivity in DDAH2 and DUSP1 genes, respectively. Promoter hypermethylation of p16 gene was found significantly associated with tumour site of buccal, gum, tongue and lip (P=0.001). In addition, DDAH2 methylation level was correlated significantly with patients' age (P=0.050). In this study, overall five-year survival rate was 38.1% for OSCC patients and was influenced by sex difference.
Conclusions: The study has identified 33 promoter hypermethylated genes that were significantly silenced in OSCC, which might be involved in an important mechanism in oral carcinogenesis. Our approaches revealed signature candidates of differentially hypermethylated genes of DDAH2 and DUSP1 which can be further developed as potential biomarkers for OSCC as diagnostic, prognostic and therapeutic targets in the future.
Keywords: DNA methylation profiling, promoter hypermethylation, Oral Squamous Cell Carcinoma, DDAH2, DUSP1, protein expression.
Introduction
Oral cancer is ranked sixth in the world's list of cancer deaths [1] and estimation of 128 000 deaths worldwide are due to oral cavity cancer in year of 2008 by the global burden of cancer (GLOBOCAN) [2]. Oral squamous cell carcinoma (OSCC) is the most prevalent type of oral cancer in the countries of the Association of Southeast Asian Nations (ASEAN) [3]. Despite the rapid advancement in multimodality treatment, the overall 5 years survival rate of OSCC has not improved much [2]. In the past decade, the cancer treatment modality and patients' prognostication depended on conventional assessment in disease staging [4]. This is not always an effective treatment strategy for halting the progress of OSCC, if the precise molecular mechanisms involved in regulating genetic and epigenetic factors remain unclear. In view of epigenetic mechanisms, DNA methylation, in particular, has provided a better understanding of cancer pathogenesis. Thus, novel and promising molecular epigenetic biomarkers for OSCC diagnosis and prognosis are continuously emerging in this area, requiring further evaluation [5, 6]. Therefore, identification of epigenetic biomarkers that are strongly associated with OSCC pathophysiology is urgently needed for early diagnostic, detection and prediction of progression, and applied molecular-targeted therapies to improve the outcomes of therapeutic treatment.
The major etiological factors for developing OSCC are smoking, alcohol consumption, human papilloma virus (HPV) and betel-quid chewing in Asian region [7, 8]. How these factors influence DNA methylation in oral carcinogenesis have not been well studied. However, smoking has been identified as a risk factor associated with hypermethylation of p16, RAS association domain family (RASSF1A) , RARβ, CDH13, MGMT and GSTP1, adenomatous polyposis coli (APC), and DNA methyltransferase 1 which catalyzes methylation process in lung cancer [9, 10]. In addition, the correlations of gene's hypermethylation with demographic and clinicopathological characteristics are known to be important for prognosis and cancer risk assessment and therapeutic effects of OSCC [11] Thus, we conducted an analysis on the correlation between hypermethylation genes with demographic and clinicopathologic characteristics of OSCC in this study.
Carcinogenesis is a multistep process, characterized by the accumulation of genetic and/or epigenetic alterations. Methylation profiling of tumour tissues helped to identify the epigenetic alterations, especially the ones involving genome-wide hypomethylation and gene-specific hypermethylation which have now been recognized as important components of carcinogenesis [12, 13]. Changes in DNA methylation have been reported to occur early in carcinogenesis and therefore are potentially important as early indicators of cancer. DNA methylation may provide an alternate pathway to gene deletion or mutation for the loss of tumour suppressor gene function [12,14, 15]. Cancer-induced silencing of hypermethylation in promoter regions of genes is important for cancer prevention and therapeutic interventions as methylated loci are reversible. Thus, identification of epigenetic biomarkers of oral cancer emerges in improving patients' survival rate.
Hypermethylation in tumour suppressor genes has been reported in various diseases including cancers. The promoter hypermethylation is one of the mechanisms leading to gene silencing by either physically inhibiting the binding of transcription factors, or by recruiting proteins that have transcription repressive properties in the cancer pathogenesis [16-18]. Thus, this epigenetic process may act as a native to genetic alterations involving DNA mutations or chromosomal aberrations that disrupt functions of the oncogenes or tumour suppressor genes [12,19, 20]. Many investigations conducted on cancer-associated genes including cyclin-dependent kinase inhibitor 2A (p16), DAPK1, RARβ, E-CADHERIN, CYCLIN A1, p14, p15, p73, RASSF1A, and APC genes, have been found to harbor hypermethylated regulatory sequences that lead to gene silencing in various cancers [5, 6, 16, 18, 21, 22]. However, no gene studied to date has been identified as a gene-specific prognostic or diagnostic biomarker in oral cancer. Thus, the identification of biomarkers for early diagnosis or as therapeutic target in oral cancer treatment is urgently needed. Dual Specificity Phosphatase 1 (DUSP1) and Dimethylarginine Dimethylamino hydrolase (DDAH2) genes are selected in this study as their relevance to OSCC progressions in differential hypermethylation of promoter regions are not clearly documented [23-25].
The recent advances in high throughput technology like microarray make it possible to generate genome-wide quantitative methylation data and identify putative candidate genes [26, 27]. In addition, various methylation dependent pre-treatments were developed to reveal the methylation status of cytosine residues, including methylation-specific polymerase chain (MS-PCR) technique. MS-PCR that utilizes sodium bisulfite treatment of genomic DNA to distinguish methylated from unmethylated cytosines, is very sensitive to detect one methylated cytosine in 1000, and the primers used are highly specific and cost effective [25, 28].
In the present study, we screened and identified differential promoter hypermethylated genes using methylation microarray analysis, and validated the methylation status by MS-PCR and protein expression by immunohistochemistry (IHC) assays. Protein expression of DDAH2 and DUSP1 has been found to be reduced in this study; therefore we presume that this is the consequence of gene expression silenced by promoter hypermethylation. In addition, the correlations of gene's hypermethylation with demographic and clinicopathological characteristics of the study are known to be important for cancer prognosis and risk assessment [11].
Materials and Methods
Samples
Twenty samples of frozen sections of primary oral squamous cell carcinoma comprising four of stage 1, five of stage 2, five of stage 3 and six of stage 4, and four samples from normal subjects with normal oral epithelial surface of reactive epithelial lesions were selected for microarray assay. Independent samples of archival Formalin-Fixed Parafin-Embedded (FFPE) of 4 normal oral mucosa of healthy subjects and 40 independent OSCC tissues were further selected for differential gene hypermethylation status validation using MS-PCR and tissue microarray consisting of 40 OSCC tissue cores for protein expression by immunohistochemical analysis. All the samples were obtained from the Malaysian Oral Cancer Database and Tumour Bank System (MOCDTBS) coordinated by Oral Cancer Research and Coordinating Center (OCRCC) of University of Malaya. Individuals' consent was obtained following appropriate Medical Ethics Committee approval of Faculty of Dentistry, University of Malaya (Medical Ethical Code no: DF OP1101/0049 (L)). No node metastases were diagnosed in all OSCC cases. Histopathological studies were confirmed by an expert oral pathologist to ensure that the samples used for DNA extraction in MS-PCR assay and IHC have more than 70% of squamous cell carcinomas tissues. All selected patients were new cases and had not been previously undergone any anticancer therapy.
DNA extraction of tissues
Genomic DNA was extracted using the DNEasy Blood and Tissue kit (Qiagen, Germany) according to the manufacturer's instructions. The quality and purity of the extracted DNA was measured using a spectrophotometer (Nanodrop; Thermo Fisher Scientific Inc., Wilmington, DE, USA). The DNA purity of the samples, measured as 260/280 ratio, had an average ratio of 1.89 ± 0.18.
Bisulphite treated DNA
The protocol was carried out according the manufacturer's instruction (EpiTect, QIAGEN GmbH, Hilden, Germany). Briefly, 1 ng - 2 ug of genomic DNA was incubated with a sodium bisulfite buffer in a thermocycler with alternating cycles of 95°C and 60°C, through steps of denaturation, sulphonation and deamination of cytosine to uracil. The resultant DNA was bound again to a membrane and then washed and incubated with a desulphonation buffer and eluted with 20 µl of elution buffer. The quality and purity of the treated DNA was measured using a spectrophotometer (Nanodrop Technologies, USA). The yields of DNA after bisulphite conversion were in the range of 50-100 ng/µl.
Methylation microarray assay
Two hundred ng of bisulfite treated DNA was applied on the Illumina Infinium HumanMethylation450 (Illumina Inc. CA, USA) following the manufacturer's protocol. Each point of methylation data was presented by fluorescent signals from the methylated (M) and unmethylated (U) alleles. Methylation status of CpG site was measured based on the ratio of fluorescent signals from one allele relative to the sum of both methylated and unmethylated alleles (β value) using Partek Genomic Suite (Partek Inc., St Loius, MO, USA) [29].
Methylation-specific PCR
The protocol for MS-PCR was performed according to the manufacturer's instructions. Briefly, the genomic DNA was converted from unmethylated cytosine to uracil by using the Epitect Bisulfite kit (Qiagen, Germany) which would not affect the methylated cytosine. Primers were designed using the Methyl Primer Express Software v1.0 (Applied Biosystems, USA). Details of the primers for unmethylated and methylated gene promoter regions are shown in Table 1. Unmethylated and methylated DNA controls (ZYMO, USA) were used as negative and positive controls, respectively. MS-PCR was performed in the Eppendorf Mastercycler Gradient PCR (Germany) with the following cycling conditions: Activation step: 95oC for 10 min.; 35 cycles of PCR in denaturation step: 94oC for 15 sec.; annealing step: Tm of primer; extension step: 72oC for 30 sec. and final extension: 72oC for 10 min. Five µl of PCR products were loaded into 2% agarose gels, electrophoresed and visualized using the Image Analyzer (Bio-Rad, Hercules, CA), after staining with SYBR Safe DNA gel stain.
MS-PCR primers and conditions
| Genes/ Conditions | P16 | DDAH2 | DUSP1 |
|---|---|---|---|
| Methylated | |||
| a) Forward primer | 5′TTATTAGAGGGTGGGGCGGATCGC3' | 5'TTGGATTACGGTCGTGTC 3' | 5'AGTTTGGAAAATTAAAGGAGC 3' |
| b) Reverse primer | 5′GACCCCGAACCGCGCCGTAA 3′ | 5'ACGAAAACTAACCTTCCCG 3' | 5'ATACCCACGTTACCTCCATA 3' |
| Unmethylated | |||
| a) Forward primer | 5′TTATTAGAGGGTGGGGTGGATGT 3′ | 5'TTTTTGGATTATGGTTGTGTT 3' | 5'GGAGTTTGGAAAATTAAAGGAGT 3' |
| b) Reverse primer | 5′CAACCCCAAACCACAACCATA A 3′ | 5'ATACAAAAACTAACCTTCCCAC 3' | 5'CAATACCCACATTACCTCCATA 3 |
| Annealing temperature (oC) | 61 | 57 | 58 |
| Product length (bp) | 152 | 112 | 119 |
| UCSC reference location | chr9:21967751-21994490 | chr6:31695894-31698245 | chr5:172197482-172199606 |
Immunohistochemical staining
Four micron-thickness of sections were deparaffined, rehydrated and subjected to antigen retrieval procedure prior to immunohistochemical (IHC) staining. Endogenous peroxidase activity was blocked by being incubated with 0.3 % H2O2. The sections were incubated with rabbit polyclonal antibodies against DDAH2 and DUSP1 (Abcam, USA) proteins. These sections were then incubated with biotinylated secondary antibody for 30 min. The antigen-antibody complexes were visualized using streptavidin-horseradish peroxidase conjugate and diaminobenzidine as chromogen. Negative controls were included by omitting the primary antibody. Slides were analyzed under a light microscope equipped with a digital camera. The IHC labeling was assessed by an experienced pathologist using staining scoring protocol as previously described [30]. All tumoural fields were evaluated for staining intensity and adjacent normal epithelium served as an internal control. Staining intensity was applied rather than counting the positively stained cells as methylation does not involve whole tissue change. The evaluation was done qualitatively by taking the definite positive brown staining at the specific cellular location of each protein. DDAH2 and DUSP1 expressions were associated with distinct nuclear and cytoplasmic staining of epithelial cells. Staining results were examined without prior knowledge of the methylation status of MS-PCR. P16 gene was not included in this procedure as its protein expression has been well studied by many other researchers [31, 32].
Methylation profiling analysis
The array signals were uploaded to the Illumina's Genome Studio software for background normalization and filtered by β values for methylation levels using the Illumina's Genome Studio software [33]. Samples which showed fluorescence intensity with P<0.001 were included in the study. Methylation level was defined as follows: Hypermethylated and hypomethylated alleles as those having an average β value in that sample of more than 0.4 above or below the overall mean for all samples. Wilcoxon rank sum test with p value of 0.001 was corrected with 5% of false discovery rate corrections (FDR) for multiple testing correction [34]. Multiple testing corrections allow a justification of p value based on test numbers being performed. Five percent of FDR are allowed having 5% chance of 1 false positive in every 500 genes. FDR adjust the p value of 0.05 to reflect the frequency of false positive in the gene list. Differences in average beta values between the two groups are presented along with the details of methylation probes. Only selected differentially hypermethylated probes in OSCC patients passed the filtration criteria [35].
The data was then exported to Partek Genomics Suite 6.5 (Partek Inc., USA) where the differentially methylated genes between normal subjects and patients were identified. Unsupervised analysis of hierarchical clustering was obtained for distribution of normal subjects' and patients' samples. The list with significant methylated genes was generated using one-way ANOVA with P < 0.05 and fold change > 2.0 [27], which was then subjected to PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System (http://www.pantherdb.org), to determine their biological pathway that are associated with carcinogenesis.
Data analysis
The statistical analysis of association between patients' demographic and clinicopathologic characteristics and the selected genes was analyzed using Pearson Chi-square, Fisher's Exact for categorical variables and independent T-tests for continuous variables in SPSS software, version 17.0 (SPSS, Chicago, USA). Patients' demographic data included in the data analysis were age, gender, alcohol drinking and tobacco smoking and betel quid chewing habits, tumour sites, pathological stages, and tumour grading. Because of the relatively small numbers in each of the four grades of pathological stages, we pooled patients into low stage (stages I and II) or high stage (stages III and IV) groups. The correlation of protein expression's strength between DDAH2 and DUSP1 was considered weak if Pearson's r was close to 0 and strong if close to 1 with significant value of P < 0.01 (two-tailed) by using Pearson correlation in SPSS software, version 17.0 (SPSS, Chicago, USA). Kaplan-Meier and log-rank tests were used to estimate the overall survival for OSCC patients and to compare survival curves between demographic, clinicopathological data and genes hypermethylation for survival rate, respectively. The association was considered as statistically significant if P < 0.05.
Results
Methylation profiling analysis
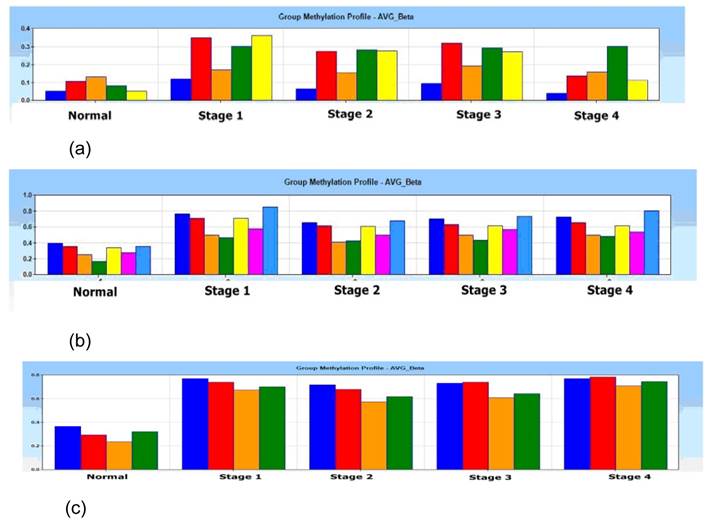
In the Illumina's Genome Studio software analysis, one of the 4 normal tissue samples was categorized and filtered as outlier as a quality control for the microarray data output, which was then excluded from the study. A gene list containing of 33 promoter-associated hypermethylated genes was generated using average β value of 0.4 in methylation (P < 0.001) (Supplementary table 1). Group methylation profiles of average beta β value for p16, DDAH2 and DUSP1 alleles were distinctly differentiated between normal and 4 pathological stages (Figs. 1 (a) - 1c)).
Histogram of group methylation profiles of (a) p16 (b) DDAH2 and (c) DUSP1 alleles average β value between normal and 4 pathological stages (Stage 1, 2, 3 and 4). Distinct profile shows lower average β value in normal subjects if compared with pathological stage 1, 2, 3 and 4 for (a) p16 (b) DDAH2 and (c) DUSP1 alleles.
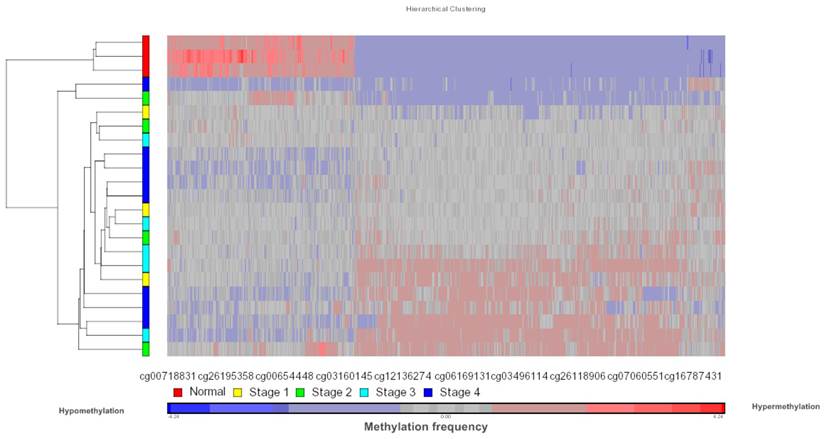
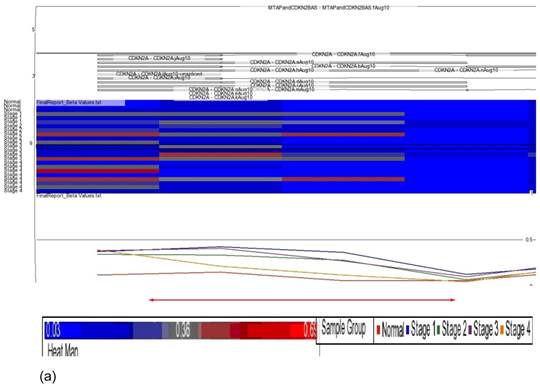
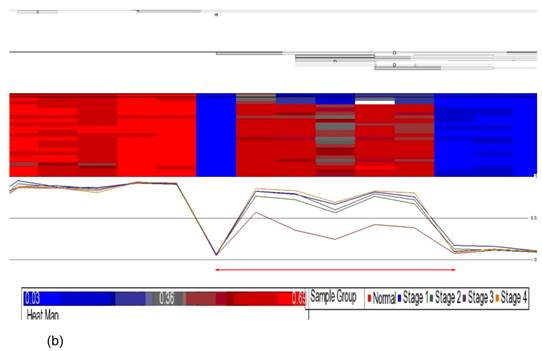
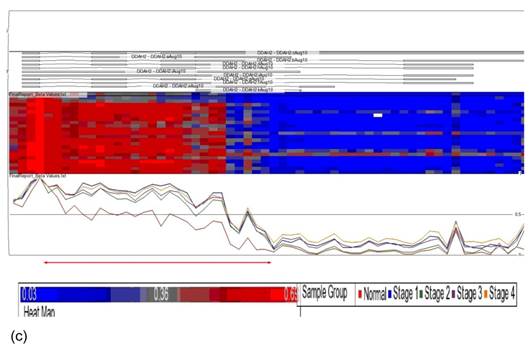
Data obtained from Illumina's Genome Studio software were analyzed by Partek Genomic Suite software. A separate heat-map hierarchical clustering analysis was performed on normal (N=3) and tumour tissues (N=20). The data set was clustered using standard hierarchical method with the Pearson's correlation to determine the distance function. The cluster set of normal and tumour samples (stage 1, 2, 3 and 4) clearly segregated the normal from tumour samples (Fig. 2). Genes that were differentially hypermethylated in their promoter regions between tumour and normal samples by at least 2 fold change and FDR value <0.05 were further mapped to the PANTHER Classification System for pathway analysis. The three significant differential genes which were selected are p16 (P= 7.94E-02), DDAH2 (P = 3.72E-01) and DUSP1 (P = 1.669e-7) for microarray data validation using MS-PCR and IHC assays. Partek Genomic Suite Visualization also demonstrated significant enrichment differences in p16, DUSP1 and DDAH2 genes for their selected probe regions respectively (P < 0.001) (Figs. 3(a) - 3(c)), which corresponded to the designed primer regions for MS-PCR analysis.
Heatmap methylation frequency of differentially methylated genes in OSCC. Unsupervised hierarchical clustering was performed on gene methylation profiles for the normal tissues (N=3) and tumour tissues (N=20, Stage 1, 2, 3 and 4). The heat map of differentially methylated genes based on clustering is shown in the figure. Each row represents a sample and each column represents a CpG loci. Red colour indicates hypermethylated CpG sites and blue colour indicates hypomethylated CpG sites.
Partek Genomic Suite Visualization. The upper panel of each shows the heat map for each probe of (a) p16, (b) DUSP1 and (c) DDAH2 genes in normal and four pathological stages. The line graphs in the lower panel of each show log2 ratio of β values of each probe between normal and four pathological stages with clear separation (indicated with red arrow).
Identification of significant differentially methylated genes
- p16: From the Partek Genomics Suite 6.5 analysis, we found that fold change of p16 was 2.134 (P=0.000). From the analysis of the Illumina's Genome Studio software, 33% of the alleles involved are located within the CpG islands, 67% in north shore of islands and all are associated with promoter regions.
- DDAH2: We decided to further investigate its methylation profile because of the observed methylation level enrichment of 2.826 fold in the study (P=0.000). From the Illumina's Genome Studio software analysis, all the DDAH2 alleles are found located within the CpG islands and 57% are involved in promoter regions; suggesting that this gene might play a role in oral cancer progression.
- DUSP1: We found that the fold change of DUSP1 was 2.566 (P=0.000). From the Illumina's Genome Studio software analysis, the DUSP1 alleles are located within the north shore of CpG islands and 25% are found in promoter regions. Further investigation of DUSP1 methylation is needed to clarify its role in oral cancer progression.
Methylation-specific PCR (MS-PCR)
These 3 selected genes were validated using MS-PCR using 4 normal tissues and 40 independent OSCC samples. The normal tissues revealed negative in methylation levels in p16, DDAH2, and DUSP1 genes. However in OSCC samples, 78%, 80%, and 88% were hypermethylated respectively for p16, DDAH2, and DUSP1 in their investigated promoter sites (Fig. 4), which corresponded with the microarray data. One hundred percent (100%) of the tumour revealed hypermethylation of the promoter region of at least one of the genes analyzed; 42.5% hypermethylation in two of the genes analyzed, and 55% hypermethylation in all of the three studied genes. There was no significant correlation among the hypermethylation status of these genes (P > 0.05).
Three representative agarose gel electrophoretic images of hypermethylation status for genes of (a) p16, (b) DDAH2 and (c) DUSP1 in methylation control and tumour samples. DKO represents methylation control; M represents methylated alleles and U represents unmethylated alleles. DKO controls (DKO M and DKO U) show bands on each gel and each tumour samples show methylated status of p16 (a), DDAH2 (b), and DUSP1 (c).

Immunohistochemical analysis
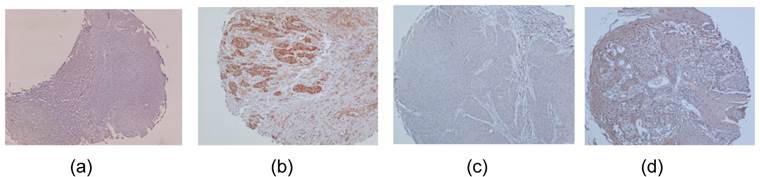
To detect protein expression levels of DUSP1 and DDAH2, IHC analysis was performed on 4 normal oral mucosa and 40 OSCC tissues. DDAH2 and DUSP1 were nuclear (Fig. 5b) and cytoplasmic (Fig. 5d) immunoreactivity. The negative controls revealed negative for immunoreactivity of DDAH2 and DUSP1 and normal mucosa showed positive in DDAH2 and DUSP1 immunoreactivity. Immunoreactivity of DDAH2 and DUSP1 was demonstrated in 24% and 22% of tumour tissues, respectively. The relationship between protein expression of DDAH2 and DUSP1 revealed a strong positive correlation (r = 0.755, P = 0.001) between these two proteins
Formaline-fixed paraffin-embedded sections of representative Oral Squamous Cell Carcinoma tissues were stained with the antibodies against DDAH2 and DUSP1. (a) Negative immunostaining of DDAH2 in nuclei of the tumour cells. (b) Positive staining of DDAH2 was detected in nuclei of the tumour cells. (c) Negative cytoplasmic immunostaining of DUSP1 in the tumour cells. (d) Positive DUSP1 cytoplasmic staining was detected in the tumour cells.
Demographic and clinicopathological characteristics of OSCC
The demographic and clinicopathologic characteristics of the 40 patients with OSCC are summarized in Table 2. These characteristics were age, gender, ethnic group, habits of alcohol drinking, tobacco smoking and betel-quid chewing, tumour sites, tumour pathological stages, invasive front and tumour grading. In survival rate data, there were two missing data or lost contact, the follow-up time for 38 patients' OSCC-related death ranged from 2 to 87 weeks, with a median follow-up time of 28.5 weeks. The ethnic groups of 40 patients consisting of 26 women and 14 men, with a median age of 59.3 years (age range of 28-80 years) were Malays (n=5), Chinese (n=6) and Indians (n=29). Most of the patients (77.5%) have habits of alcohol drinking, tobacco smoking and/or betel-quid chewing while only 20% of the patients have none of the aforementioned habits. For tumour site, 19 (47.5%) were from buccal mucosa, 8 (20%) gum, 12 (30%) tongue and 1 (2.5%) lip. For pathological stages, one patient (2.5%) had TNM stage 0 disease, 4 (10%) stage I, 6 (15%) stage II, 3 (7.5%) stage III, and 26 (65%) stage IV. For invasive front, four (10%) cases each for negative and cohensive front, 29 (72.5%) of non cohensive front and 3 (7.5%) of no stated cases. In tumour grading, 13 (32.5%) had well differentiation, 25 (62.5%) moderate differentiation and one (2.5%) each for poorly and no stated cases.
Demographic and clinicopathological characteristics
| Characteristics | Frequency (Percentage) | P value of methylated genes | ||
|---|---|---|---|---|
| p16 | DDAH2 | DUSP1 | ||
| Gender | ||||
| Male | 14 (35%) | |||
| Female | 26 (66%) | 0.546 | 0.168 | 0.631 |
| Ethnic | ||||
| Malay | 5 (12.5%) | |||
| Chinese | 6 (15.0%) | |||
| Indian | 29 (72.5.0%) | 0.214 | 0.473 | 0.560 |
| Age Range (Years) | ||||
| 20-39 | 4 (10%) | |||
| 40-59 | 15 (37.5%) | |||
| 60-79 | 19 (47.5%) | |||
| > 80 | 2 (5%) | 0.196 | 0.050* | 0.574 |
| Habit | ||||
| No habit | 8 (20%) | |||
| Quit habit | 1 (2.5%) | |||
| Alcohol drinking/Tobacco smoking/Betel-quid chewing | 31 (77.5%) | 0.855 | 0.541 | 0.575 |
| Tumors Site | ||||
| Buccal mucosa | 19 (47.5%) | |||
| Gum | 8(20%) | |||
| Tongue | 12 (30%) | |||
| Lip | 1 (2.5%) | 0.001* | 0.700 | 0.601 |
| Pathological TNM stage | ||||
| I | 4 (10%) | |||
| II | 6 (15%) | |||
| III | 4 (10%) | |||
| IV | 26 (65%) | 0.327 | 0.587 | 0.325 |
| Invasive front (POI staging) | ||||
| Cohensive | 6 (15%) | |||
| Non cohensive | 31 (77.5%) | |||
| No stated | 3 (7.5%) | 0.247 | 0.542 | 0.278 |
| Differentiation (Tumour grading) | ||||
| Well | 13 (32.5%) | |||
| Moderate | 25 (62.5%) | |||
| Poorly | 1 (2.5%) | |||
| No stated | 1 (2.5%) | 0.862 | 0.858 | 0.735 |
*Significant difference for promoter hypermethylation in p16 with tumour site by Fisher's Exact test (P=0.001), and DDAH2 with patients' age by independent-samples T test (P=0.050).
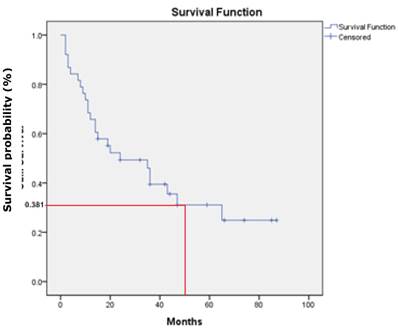
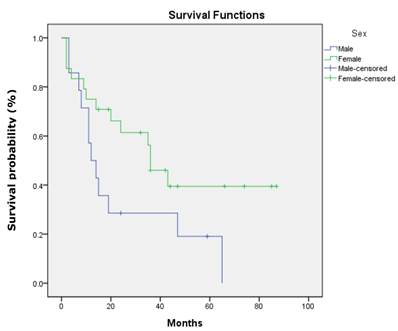
There was no statistical significant association between the three studied genes with demographic and clinicopathologic characteristic of patients' age, gender, ethnicity, habits, tumour sites, pathological stages, invasive front and tumour grading (P>.0.05). However, a statistically significant association was found between p16 gene promoter region with tumour site of buccal, gum, tongue and lip (P=0.001). In addition, DDAH2 hypermethylation was correlated significantly with patients' age (P=0.050). In this study, the overall survival rate showed 38.1% for OSCC patients (95% CI=27.1, 49.1) (Fig. 6) and there was a significant difference in the survival rate percentage with 24.2% for male and 46.5% for female in the sex difference (P=0.050) (Fig.7). However, patients' age, ethnicity, habits, tumour sites, tumour pathological stages, invasive front and tumour grading, and hypermethylation of p16, DUSP1 and DDAH2 did not influence the survival rate (P>0.05).
Kaplan-Meier survival curve shows 38.1% of overall five-year survival rate.
Kaplan-Meier survival curve shows sex difference influences differently on OSCC patients' overall five-year survival rate.
Discussions
In our study, the methylation profiling demonstrated clear segregated pattern of the normal from tumour samples, which was concordant with those reported elsewhere [33, 36, 37]. The microarray-based methylation profiling was very specific and useful for the identification of signature gene and genes that are involved in oral carcinogenesis [38], as the microarray reliability has been previously demonstrated and validated with single gene assays by Bibikova et al [29] and very limited methylation array data available in OSCC. To the best of our knowledge, this is the first DNA methylation profiling on Malaysian oral cancer using the Illumina, Infinium Methylation 450K assay. This approach provided comprehensive and high throughput data for a genome wide study, which covered more than 450,000 methylation loci per sample at single nucleotide resolution. Further validation of candidate genes on a separate cohort of healthy and oral cancer population by MS-PCR analysis has allowed us to confirm hypermethylation in promoter regions of p16 and DUSP1 as reported by others [39, 40]. Thus, we confirmed the gene methylation status using methylation specific technique. MS-PCR and IHC analysis in DUSP1 and DDAH2 genes were reliable as the microarray data showed 100% concordance with other studies [41, 42]. Therefore, using computational and gene-specific validation approaches, we were able to identify the potential epigenetic biomarkers of oral cancer.
Tumour suppressor gene, p16 (chromosome 9p21.3) is a cell cycle regulator involved in the inhibition of G1 phase progression in normal cells. However, promoter hypermethylation of p16 gene in CpG islands silences its transcription and represses gene expression. In our study, this gene with promoter CpG island hypermethylation associated with transcriptional silencing, is an early frequent event in multiple human cancers including lung, prostate, breast and OSCC [14, 43-45]. The inverse correlation between the p16 gene hypermethylation and immunoreactivity suggests that hypermethylation mechanism is an important event for p16 gene aberration in various cancers [31, 32, 46].
DDAH2 gene (chromosome 6p21.3) encodes an enzyme that is involved in nitric oxide (NO) generation by regulating cellular concentrations of methyl arginine, which in turn inhibits nitric oxide synthase (NOS) activity in normal cells [47]. NO is involved in vital cell processes of vasodilation, respiration, cell migration, immune response and apoptosis. However, its dysregulation has been implicated in many pathophysiological conditions such as chronic disease and cancer [24, 48-51], including the OSCC cases in our study. In this study, DDAH2 gene was found as a candidate of hypermethylated gene with down-regulation of protein expression in OSCC, which seems to play an important role in cancer progression. There is a controversy with a recent finding reported by Kim et al (2010) showing that the up-regulation of DDAH2 gene expression, which is involved in the apoptosis inhibitory process, might be associated with ovarian carcinoma poor prognosis [49].
The DUSP1 gene (chromosome 5q35.1) encodes for the dual specificity protein phosphatase enzyme. It acts as an activator in MAPK signaling pathway, to dephosphorylate MAP kinase (ERK-2) on both 'Thr-183' and 'Tyr-185' which are actively involved in protein modification, signal transduction and oxidative stress in normal cells. In addition, DUSP1 is controlled by p53 during cellular response to oxidative stress. It is also involved in angiogenesis, invasion and metastasis of non-small cell lung cancer [52]. High level of DUSP1 promoter hypermethylation was demonstrated in our tumour samples as have been reported by other cancer researches [39, 52, 53].
In the study, the normal tissues did not show hypermethylation levels; vice-versa in OSCC tissues, there are hypermethylated evidences in p16, DDAH2 and DUSP1 genes. These gene promoters are not methylated during the germ line, however, dysregulation of methylation of these gene promoters that silences the gene expression causing the cancer progression [43, 49, 50, 52]. We found no correlation of the hypermethylation status among any of the p16, DDAH2 and DUSP1 genes. This indicates that the hypermethylation of these promoter regions are gene specific of OSCC in this study. The association of gene hypermethylation with OSCC clinicopathological characteristics is essential for better understanding of how hypermethylated genes are involved in OSCC development. The findings showed no correlation between demographic and clinicopathological characteristics of OSCC patients' with the genes' hypermethylation, except p16, which was statistically significant as it was associated with tumour site of buccal, gum, tongue, and lip (P = 0.001) and DDAH2 hypermethylation level was correlated significantly with patients' age (P=0.050). The overall five-year survival rate reported by Uğurluer et al (2006) was 39% [54], which is very similar to our findings. Our study revealed no influence of demographic and clinicopathological characteristics, and DUSP1 and DDAH2 immunoexpression (p>0.05) except sex difference (P=0.050) on OSCC patients' survival rate. A large population-based study conducted by Majek et al (2013) confirmed a survival advantage of sex difference on colorectal cancer patients and this is concordant with the finding of our study [55].
Our finding revealed that p16 gene hypermethylated differentially in various tumour sites of the oral cavity. Therefore, cancer cells seem to harbor a heterogeneous epigenetic alteration caused by a series of clonal expansion of p16 genetic changes in various tumour sites of the oral cavity [56]. The studies conducted on different HNSCC tumour sites [13] and HPV-driven SCC cases [57] revealed different correlative significant levels of gene hypermethylation. The lack of significant correlative association between the genes' promoter hypermethylation with demographical and clinicopathological characteristics is most probably due to the limited number of samples employed in our study. Therefore, we need a larger OSCC sample size for further studies to determine a significant hypermethylation status that could be associated with clinical outcomes.
Expressions of the gene products were examined by immunohistochemistry (IHC). In the IHC study, the inverse correlation of hypermethylation level of DDAH2 and DUSP1 and protein expression was evidenced in primary OSCC tissues, suggesting that protein expression of DDAH2 and DUSP1 was silenced by promoter hypermethylation in OSCC. Promoter hypermethylation is an epigenetic change which acts as one of the pathways leadings to oral carcinogenesis [14]. Similar finding about DUSP1 hypermethylation in hepatocellular carcinoma has been reported by Calvisi et al [53]. However, to the best of our knowledge, presently there are no other cancer reports about the effect of DDAH2 hypermethylation on its protein expression. In the study, the increase of protein expression of the DDAH2 does significantly relate to increase of DUSP1, which means that both proteins are most likely expressed together in the OSCC cases.
In conclusions, our promoter hypermethylation biomarkers screening approach with microarray technology and validation by MS-PCR and IHC analysis had identified specific candidate genes that are DUSP1 and DDAH2 genes for OSCC. Such novel biomarkers would be useful for diagnostic, prognostic or therapeutic applications. The methylation profiling to identify differential promoter hypermethylation between OSCC and normal samples may provide a tool to decode the molecular understanding in OSCC progression. Our results indicate that protein expression of DDAH2 and DUSP1 genes were reduced by their promoter hypermethylation in OSCC, as promoter hypermethylation is the predominant mechanism in DDAH2 and DUSP1 deregulation. Further work will be aimed at elucidating the functional roles of DDAH2 and DUSP1 in larger OSCC samples for their potential use as hypermethylated-based biomarkers.
Supplementary Material
Supplementary Table 1Gene list of promoter-associated hypermethylated genes of OSCC generated by Illumina's Genome Studio software analysis.
Acknowledgements
This work was supported by the PPP Grant (PV046-2011) of University of Malaya, Malaysia, the Research Intensive Faculty (600-RMI/DANA 5/3/RIF (633/2012)) grant of Universiti Teknologi MARA, Malaysia, Science fund grant of Ministry of Science, Technology and innovation (MOSTI), Malaysia and the Fundamental Research Grant Scheme (FRGS) of the Ministry of Higher Education, Malaysia. We thank for the Oral Cancer Research and Coordinating Centre (OCRCC), University of Malaya for providing the tissues and data from the Malaysia Oral Cancer Database and Tissue bank System (MOCDTBS). We acknowledge the Institute of Medical Molecular Biotechnology (IMMB), Faculty of Medicine, Universiti Teknologi MARA, Selangor, Malaysia for providing laboratory facilities.
Competing interests
The authors have declared that no competing interest.
References
1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA: a cancer journal for clinicians. 2005;55:74-108
2. Jemal A, Bray F, Center MM. et al. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61:69-90
3. Kimman M, Norman R, Jan S. et al. The burden of cancer in member countries of the Association of Southeast Asian Nations (ASEAN). Asian Pac J Cancer Prev. 2012;13:411-420
4. Kim TJ, Kim HY, Lee KW, Kim MS. Multimodality Assessment of Esophageal Cancer: Preoperative Staging and Monitoring of Response to Therapy. Radiographics. 2009;29:403-421
5. Mulero-Navarro S, Esteller M. Epigenetic biomarkers for human cancer: The time is now. Critical Reviews in Oncology/Hematology. 2008;68:1-11
6. Balgkouranidou I, Liloglou T, Lianidou ES. Lung cancer epigenetics: emerging biomarkers. Biomarkers in Medicine. 2013;7:49-58
7. Liu SA. A Literature Analysis of the Risk Factors for Oral Cancer. Book chapter. 2012
8. Goot-Heah K, Kwai-Lin T, Froemming GRA. et al. Human Papilloma Virus 18 Detection in Oral Squamous Cell Carcinoma and Potentially Malignant Lesions Using Saliva Samples. Asian Pacific Journal of Cancer Prevention. 2012;13:6109-6113
9. Toyooka S, Maruyama R, Toyooka KO. et al. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. International Journal of Cancer. 2002;103:153-160
10. Lin RK, Hsieh YS, Lin P. et al. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. The Journal of clinical investigation. 2010;120:521-532
11. Maruyama R, Toyooka S, Toyooka KO. et al. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clinical cancer research. 2002;8:514-519
12. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nature Reviews Cancer. 2006;6:107-116
13. Poage GM, Houseman EA, Christensen BC. et al. Global hypomethylation identifies loci targeted for hypermethylation in head and neck cancer. Clinical cancer research. 2011;17:3579-3589
14. Ha PK, Califano JA. Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. The lancet oncology. 2006;7:77-82
15. Khor GH, Froemming GA, Tan AC. et al. Pathways Deregulation in Oral Squamous Cell Carcinoma using Methylation Profiling. Journal Dental Research. 2011;91(Special issue C):abs47
16. Miyamoto K, Ushijima T. Diagnostic and Therapeutic Applications of Epigenetics. Japanese Journal of Clinical Oncology. 2005;35:293-301
17. Baylin SB. DNA methylation and gene silencing in cancer. Nature Clinical Practice Oncology. 2005;2:S4-S11
18. Radhakrishnan R, Kabekkodu S, Satyamoorthy K. DNA hypermethylation as an epigenetic mark for oral cancer diagnosis. Journal of Oral Pathology & Medicine. 2011;40:665-676
19. Khor GH, Hassan MI, Siar CH. p53 Expression as a marker of microinvasion in oral squamous cell carcinoma. Asian Pac J Cancer Prev. 2011;12:1017-1022
20. Khor GH. Chromosome 17 Aberration of Oral Squamous Cell Carcinoma in Malaysia. Global Jof Health Sci. 2009. 2009;1:150-156
21. Shaw R. The epigenetics of oral cancer. International Journal of Oral and Maxillofacial Surgery. 2006;35:101-108
22. Ovchinnikov DA, Cooper MA, Pandit P. et al. Tumor-suppressor Gene Promoter Hypermethylation in Saliva of Head and Neck Cancer Patients. Translational oncology. 2012;5:321-326
23. Buytaert E, Matroule J, Durinck S. et al. Molecular effectors and modulators of hypericin-mediated cell death in bladder cancer cells. Oncogene. 2007;27:1916-1929
24. Zhang J, Liu J, Li Z. et al. Dysfunction of endothelial NO system originated from homocysteine-induced aberrant methylation pattern in promoter region of DDAH2 gene. Chinese Medical Journal. 2007;120:2132-2137
25. Galm O, Herman JG. Methylation-specific polymerase chain reaction. Methods Mol Med. 2005;113:279-291
26. Gitan RS, Shi H, Chen CM. et al. Methylation-specific oligonucleotide microarray: a new potential for high-throughput methylation analysis. Genome research. 2002;12:158-164
27. Kron K, Pethe V, Briollais L. et al. Discovery of novel hypermethylated genes in prostate cancer using genomic CpG island microarrays. PloS one. 2009;4:e4830
28. Wong IHN. Qualitative and quantitative polymerase chain reaction-based methods for DNA methylation analyses. Methods in Molecular Biology. 2006;336:33-43
29. Bibikova M, Barnes B, Tsan C. et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288-295
30. Van Diest P, Van Dam P, Henzen-Logmans S. et al. A scoring system for immunohistochemical staining: consensus report of the task force for basic research of the EORTC-GCCG. European Organization for Research and Treatment of Cancer-Gynaecological Cancer Cooperative Group. Journal of clinical pathology. 1997;50:801-804
31. Shim YH, Kang GH, Ro JY. Correlation of p16 hypermethylation with p16 protein loss in sporadic gastric carcinomas. Laboratory investigation. 2000;80:689-695
32. Klangby U, Okan I, Magnusson KP. et al. p16/INK4a and p15/INK4b gene methylation and absence of p16/INK4a mRNA and protein expression in Burkitt's lymphoma. Blood. 1998;91:1680-1687
33. Bibikova M, Barnes B, Tsan C. et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288-295
34. Olson NE. The microarray data analysis process: from raw data to biological significance. NeuroRx. 2006;3:373-383
35. Bibikova M, Le J, Barnes B. et al. Genome-wide DNA methylation profiling using Infinium assay. Epigenomics. 2009;1:177-200
36. Killian JK, Walker RL, Bilke S. et al. Genome-Wide Methylation Profiling in Archival Formalin-Fixed Paraffin-Embedded Tissue Samples. Methods Mol Biol. 2012;823:107-118
37. Jithesh P, Risk J, Schache A. et al. The epigenetic landscape of oral squamous cell carcinoma. British journal of cancer. 2013;108:370-379
38. Nazmul-Hossain A, Patel K, Rhodus N, Moser K. Microarrays: applications in dental research. Oral Diseases. 2008;14:25-29
39. Rauhala HE, Porkka KP, Tolonen TT. et al. Dual-specificity phosphatase 1 and serum/glucocorticoid-regulated kinase are downregulated in prostate cancer. International Journal of Cancer. 2005;117:738-745
40. Cao J, Zhou J, Gao Y. et al. Methylation of p16 CpG Island Associated with Malignant Progression of Oral Epithelial Dysplasia: A Prospective Cohort Study. Clinical cancer research. 2009;15:5178-5183
41. Weber M, Davies JJ, Wittig D. et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature genetics. 2005;37:853-862
42. Bibikova M, Lin ZW, Zhou LX. et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Research. 2006;16:383-393
43. Shaw RJ, Liloglou T, Rogers SN. et al. Promoter methylation of P16, RARβ, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. British journal of cancer. 2006;94:561-568
44. Sharma G, Mirza S, Prasad CP. et al. Promoter hypermethylation of p16 INK4A,p14,ARF,CyclinD2 and Slit2 in serum and tumor DNA from breast cancer patients. Life sciences. 2007;80:1873-1881
45. Jabłonowski Z, Reszka E, Gromadzińska J. et al. Hypermethylation of p16 and DAPK promoter gene regions in patients with non-invasive urinary bladder cancer. Arch Med Sci. 2011;7:512-516
46. Milde-Langosch K, Ocon E, Becker G, Löning T. p16/MTS1 inactivation in ovarian carcinomas: High frequency of reduced protein expression associated with hyper-methylation or mutation in endometrioid and mucinous tumors. International Journal of Cancer. 1998:61-65
47. Leiper J, Nandi M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nature Reviews Drug Discovery. 2011;10:277-291
48. Hasegawa K, Wakino S, Tanaka T. et al. Dimethylarginine dimethylaminohydrolase 2 increases vascular endothelial growth factor expression through Sp1 transcription factor in endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1488-1494
49. Kim YS, Do JH, Bae S. et al. Identification of differentially expressed genes using an annealing control primer system in stage III serous ovarian carcinoma. BMC cancer. 2010;10:576-590
50. Korde S, Sridharan G, Gadbail A, Poornima V. Nitric oxide and oral cancer: A review. Oral Oncology. 2012;48:475-483
51. Abhary S, Burdon KP, Kuot A. et al. Sequence variation in DDAH1 and DDAH2 genes is strongly and additively associated with serum ADMA concentrations in individuals with type 2 diabetes. PLoS One. 2010;5:1-8
52. Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer and Metastasis Reviews. 2008;27:253-261
53. Calvisi DF, Pinna F, Meloni F. et al. Dual-Specificity Phosphatase 1 Ubiquitination in Extracellular Signal-Regulated Kinase-Mediated Control of Growth in Human Hepatocellular Carcinoma. Cancer research. 2008;68:4192-4200
54. Uğurluer G, Ozyurt S, Aksaray F. et al. Prognostic factors and survival rates in oral cavity cancers. Journal of ear, nose, and throat. 2006;16:64-71
55. Majek O, Gondos A, Jansen L. et al. Sex Differences in Colorectal Cancer Survival: Population-Based Analysis of 164,996 Colorectal Cancer Patients in Germany. PLoS One. 2013;8:e68077
56. Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nature reviews genetics. 2006;7:21-33
57. Sartor MA, Dolinoy DC, Jones TR. et al. Genome-wide methylation and expression differences in HPV (+) and HPV (-) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics. 2011;6:777-787
Author contact
![]() Corresponding author: Kwai Lin Thong, Institute of Biological Science, Faculty of Science, University of Malaya, 50603, Kuala Lumpur, Malaysia. E-mail: thongkledu.my. Tel: +603-79674437 Fax: +603-79675908 Or Goot Heah Khor, Centre of Studies for Preclinical Science, Faculty of Dentistry, Universiti Teknologi MARA, 40450. Shah Alam, Selangor, Malaysia. E-mail: gootheahuitm.edu.my Tel: +603-55435814 Fax: +603-55435803.
Corresponding author: Kwai Lin Thong, Institute of Biological Science, Faculty of Science, University of Malaya, 50603, Kuala Lumpur, Malaysia. E-mail: thongkledu.my. Tel: +603-79674437 Fax: +603-79675908 Or Goot Heah Khor, Centre of Studies for Preclinical Science, Faculty of Dentistry, Universiti Teknologi MARA, 40450. Shah Alam, Selangor, Malaysia. E-mail: gootheahuitm.edu.my Tel: +603-55435814 Fax: +603-55435803.