Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(10):1422-1429. doi:10.7150/ijms.6809 This issue Cite
Research Paper
PITX2c Loss-of-Function Mutations Responsible for Congenital Atrial Septal Defects
Fang Yuan1#, Lan Zhao2#, Juan Wang3, Wei Zhang4, Xin Li5, Xing-Biao Qiu1, Ruo-Gu Li1, Ying-Jia Xu1, Lei Xu1, Xing-Kai Qu1, Wei-Yi Fang1 ![]() , Yi-Qing Yang1,6
, Yi-Qing Yang1,6 ![]()
1. Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China
2. Department of Cardiology, Yantaishan Hospital, 91 Jiefang Road, Yantai 264001, Shandong, China
3. Department of Cardiology, East Hospital, Tongji University School of Medicine, 150 Jimo Road, Shanghai 200120, China
4. Department of Cardiac Surgery, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China
5. Department of Extracorporeal Circulation, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China
6. Department of Cardiovascular Research, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China
# Contributed equally to this work.
Received 2013-5-31; Accepted 2013-7-29; Published 2013-8-22
Abstract
Congenital heart disease (CHD) is the most common form of developmental anomaly and is the leading non-infectious cause of infant mortality. A growing body of evidence demonstrates that genetic risk factors are involved in the pathogenesis of CHD. However, CHD is a genetically heterogeneous disease and the genetic determinants for CHD in most patients remain unclear. In the present study, the entire coding region and splice junction sites of the PITX2c gene, which encodes a homeobox transcription factor crucial for normal cardiovascular genesis, was sequenced in 150 unrelated patients with various CHDs. The 200 unrelated control individuals were subsequently genotyped. The functional characteristics of the mutations were explored using a dual-luciferase reporter assay system. As a result, two novel heterozygous PITX2c mutations, p.H98Q and p.M119T, were identified in 2 unrelated patients with atrial septal defects, respectively. The variations were absent in 400 control chromosomes and the affected amino acids were completely conserved evolutionarily. The two variants were both predicted to be disease-causing by MutationTaster and PolyPhen-2, and the functional analysis revealed that the PITX2c mutants were consistently associated with significantly reduced transcriptional activity compared with their wild-type counterpart. These findings firstly link PITX2c loss-of-function mutations to atrial septal defects in humans, which provide novel insight into the molecular mechanism responsible for CHD, suggesting potential implications for the early prophylaxis and allele-specific treatment of CHD.
Keywords: Congenital heart disease, Atrial septal defect, Genetics, Transcription factor, PITX2c
Introduction
Congenital heart disease (CHD), also known as congenital cardiovascular defect that arises from abnormal formation of the heart or major blood vessels, is the most common form of major developmental anomaly in humans worldwide, with a prevalence of approximately 1% among live births, and is the leading non-infectious cause of infant death, with an estimated 30% of newborns who die of birth defects having cardiovascular abnormalities [1]. Congenital cardiovascular malformations are clinically classified into at least 21 different categories with specific anatomic lesions, including ventricular septal defect, atrial septal defect, atrioventricular septal defect, tetraology of Fallot, patent ductus arteriosus, transposition of the great artery, right ventricular outflow tract obstruction, aortic stenosis, coronary artery deformation, tricuspid atresia, and Ebstein's anomaly, of which ventricular septal defect is the most prevalent congenital heart defect at birth, while atrial septal defect is the most common adult congenital heart defect and tetraology of Fallot is the most common type of cyanotic congenital heart disease [1,2]. Various CHDs can occur separately or in combination, resulting in reduced exercise capacity, degraded quality of life, delayed fetal brain development, cardiac dysfunction or congestive heart failure, thromboembolic stroke, pulmonary embolus, subacute bacterial endocarditis, arrhythmias, and even sudden cardiac death [3-12]. Hence, CHD has imposed a substantial economic burden on patients and health care systems, and the socioeconomic burden is anticipated to increase in the future with increasing CHD adults [13]. Despite the pronounced morbidity and significant mortality throughout life, the molecular mechanism underlying CHD remains poorly understood.
A growing body of studies demonstrates that cardiovascular morphogenesis is a complex and dynamic biological process that requires the orchestration of cardiac cell commitment, differentiation, proliferation, and migration, with both environmental and genetic risk factors being implicated in this heterogeneous process [14-16]. Based on their temporal and spatial expression profiles and their phenotypic characteristics associated with loss or gain of function, many cardiac transcription factors are increasingly recognized as playing crucial roles in embryonic cardiogenesis, including NK and GATA families [17-21]. However, CHD is a genetically heterogeneous disease and the genetic determinants for CHD in an overwhelming majority of patients remain to be identified.
Emerging evidence indicates that the cardiac transcription factor PITX2c, a member of the pituitary homeobox (PITX) family of transcription factors, plays an important role in the normal cardiovascular genesis [22-27]. The PITX2c gene is predominantly expressed in the embryonic and adult hearts [28], and is essential for the embryonic development of left atrium, cardiac conduction system, and pulmonary venous myocardium [29]. In mice, PITX2 knockout led to embryonic lethality at mid-term, mainly due to severe cardiovascular defects, including double-outlet right ventricle, atrial isomerism, atrial septal defect, ventricular septal defect, transposition of the great artery, persistent truncus arteriosus, and abnormal aortic arch as well as incomplete closure of the body wall [30-32], and isoform-specific PITX2 deletion revealed that PITX2c was the most relevant isoform during the development of embryonic heart, since lack of PITX2c expression, but not PITX2a or PITX2b, recapitulated the morphogenetic defects similar to systemic PITX2-deficient mice [32]. In Xenopus, partial depletion of PITX2c mRNA by using chemically modified antisense oligonucleotides resulted in aberrant cardiac morphology, of which the most common cardiac abnormality seen was a failure of rightward migration of the outflow tract, present in 23% of embryos injected with the PITX2c antisense oligonucleotide. In addition, a number of other cardiac anomalies were observed after PITX2c mRNA attenuation, including abnormalities of atrial septation, extracellular matrix restriction, relative atrial-ventricular chamber positioning, and restriction of ventricular development [23]. These findings justify PITX2c as a prime candidate gene for CHD.
Materials and Methods
Study participants
A cohort of 150 unrelated patients with CHD was recruited among Chinese Han population. The patients were evaluated by individual and familial histories, review of the medical records, complete physical examination, 12-lead electrocardiogram, and two-dimensional transthoracic echocardiography with color flow Doppler. All patients had echocardiogram documented CHDs. Most patients underwent cardiac surgery or catheter-based repair. The patients with known chromosomal abnormalities or syndromic cardiovascular defects were excluded from the study.
A total of 200 unrelated, ethnically matched healthy individuals randomly selected from the individuals undergoing routine physical examinations were used as control subjects. According to reviews of medical histories and analyses of echocardiographic records, the control individuals had no CHDs, except for subclinical cardiac aberrations such as bicuspid aortic valve and patent foramen ovale. The ethnic origin of a participant was ascertained by a combination of self-reported ethnicity and a personal questionnaire asking questions about the birthplace, language, religion, and ancestry [20].
Peripheral venous blood specimens from CHD cases and control individuals were prepared. The study protocol was reviewed and approved by the local institutional ethics committee and written informed consent was obtained from all participants or their guardians prior to study.
Genotyping for PITX2c
Genomic DNA from all participants was extracted from blood lymphocytes with Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). The candidate gene PITX2c was sequenced initially in 150 unrelated patients with CHD, and genotyping PITX2c was performed subsequently in the 200 unrelated control individuals for the identified mutations in CHD patients. The referential genomic DNA sequence of PITX2c derived from GenBank (accession No. NC_000004), which was at the National Center for Biotechnical Information (NCBI; http://www.ncbi.nlm.nih.gov/). With the help of on-line Primer 3 software (http://frodo.wi.mit.edu), the primer pairs used to amplify the whole coding region and splice junction sites of PITX2c by polymerase chain reaction (PCR) were designed as previously described [33]. The PCR was performed using HotStar Taq DNA Polymerase (Qiagen GmbH, Hilden, Germany) on a PE 9700 Thermal Cycler (Applied Biosystems, Foster, CA, USA), with standard conditions and concentrations of reagents. Amplified products were analyzed on 1% agarose gels stained with ethidium bromide and purified with QIAquick Gel Extraction Kit (Qiagen). Both strands of each PCR product were sequenced with a BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) under an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems). The sequencing primers were the same as previously designed for specific region amplification. The DNA sequences were viewed and analyzed with the DNA Sequencing Analysis Software v5.1 (Applied Biosystems). The variant was validated by re-sequencing an independent PCR-generated amplicon from the subject and met the quality control thresholds with a call rate >99%. Additionally, for an identified sequence variant, the Exome Variant Server (EVS; http://evs.gs.washington.edu/EVS) and NCBI's single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) online databases were queried to confirm its novelty.
Alignment of multiple PITX2c protein sequences
Multiple PITX2c protein sequences across various species were aligned using the online program of MUSCLE, version 3.6 (http://www.ncbi.nlm.nih.gov/).
Prediction of the causative potential of a PITX2c sequence variation
The disease-causing potential of a PITX2c sequence variation was predicted by MutationTaster (an online program at http://www.mutationtaster.org), which automatically gave a probability for the variation to be either a pathogenic mutation or a benign polymorphism. Notably, the P value used here is the probability of the correct prediction rather than the probability of error as used in t test statistics (i.e., a value close to 1 indicates high accuracy of the prediction). In addition, another online program PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) was also used to evaluate the causative likeliness of a variant.
Expression plasmids and site-directed mutagenesis
The recombinant expression plasmid PITX2c-pcDNA4, which was constructed by M. Hermina Strungaru and colleagues [34], was a gift from Prof. Georges Christé, from Physiopathologie des Troubles du Rythme Cardiaque, Faculté de Pharmacie de Lyon, Université Lyon 1, Lyon, France. The atrial natriuretic factor (ANF)-luciferase reporter plasmid, which contains the 2600-bp 5'-flanking region of the ANF gene, namely ANF(-2600)-Luc, was kindly provided by Dr. Ichiro Shiojima, from the Department of Cardiovascular Science and Medicine, Chiba University Graduate School of Medicine, Chuo-ku, Chiba, Japan. The identified mutation was introduced into the wild-type PITX2c using a QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) with a complementary pair of primers. The mutant was sequenced to confirm the desired mutation and to exclude any other sequence variations.
Luciferase reporter gene assay
Chinese hamster ovary (CHO) cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum as well as 100 units/ml penicillin and 100 μg/ml streptomycin. CHO cells were cultured 24 h prior to transfection. The ANF(-2600)-Luc reporter construct and an internal control reporter plasmid pGL4.75 (hRluc/CMV, Promega) were used in transient transfection assays to explore the transactivational activity of the PITX2c mutants. CHO cells were transfected with 2 μg of wild-type PITX2c-pcDNA4 or mutant PITX2c-pcDNA4 (H98Q or M119T) or empty vector pcDNA4, 2.0 μg of ANF(-2600)-Luc reporter construct, and 0.04 μg of pGL4.75 control reporter vector using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA). For co-transfection experiments, 1 μg of wild-type PITX2c-pcDNA4, 1 μg of mutant PITX2c-pcDNA4 (H98Q or M119T), 2.0 μg of ANF(-2600)-Luc, and 0.04 μg of pGL4.75 were used. Transfected cells were incubated for 24 h, then lysed and assayed for reporter activities. Firefly luciferase and Renilla luciferase activities were measured with the Dual-Glo luciferase assay system (Promega). The activity of the ANF promoter was presented as fold activation of Firefly luciferase relative to Renilla luciferase. Three independent experiments were performed at minimum for wild-type or mutant PITX2c.
Statistical analysis
Data are expressed as means ± standard deviations. Continuous variables were tested for normality of distribution, and Student's unpaired t test was used for comparison of numeric variables between two groups. Comparison of the categorical variables between two groups was performed using Pearson's χ2 test or Fisher's exact test when appropriate. A two-tailed P value less than 0.05 was considered to be statistically significant.
Results
Baseline characteristics of the study subjects
A cohort of 150 unrelated patients with CHD was enlisted and clinically evaluated in contrast to a total of 200 unrelated, ethnically-matched healthy individuals used as controls. All the participants had no established environmental risk factors for CHD, such as maternal illness and drug use in the first trimester of pregnancy, parental smoking, and long-term exposure to toxicants and ionizing radiation. The baseline clinical characteristics of the 150 unrelated CHD patients are summarized in Table 1.
Clinical characteristics of the 150 unrelated patients with congenital heart disease (CHD).
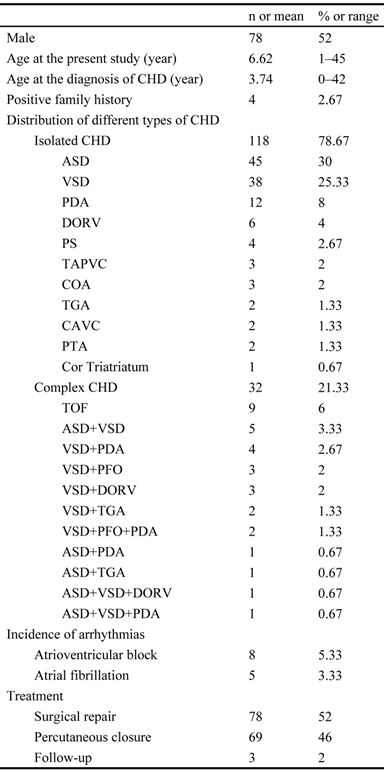
CHD: congenital heart disease, ASD: atrial septal defect, VSD: ventricular septal defect, TOF: tetralogy of Fallot, PDA: patent ductus arteriosus, DORV: double outlet right ventricle, PS: pulmonary stenosis, TAPVC: total abnormal pulmonary venous connection, COA: coarctation of the aorta, TGA: transposition of the great arteries, CAVC: common arteriovenous canal, PTA: persistent truncus arteriosus, PFO: patent foramen ovale.
PITX2c sequence variations
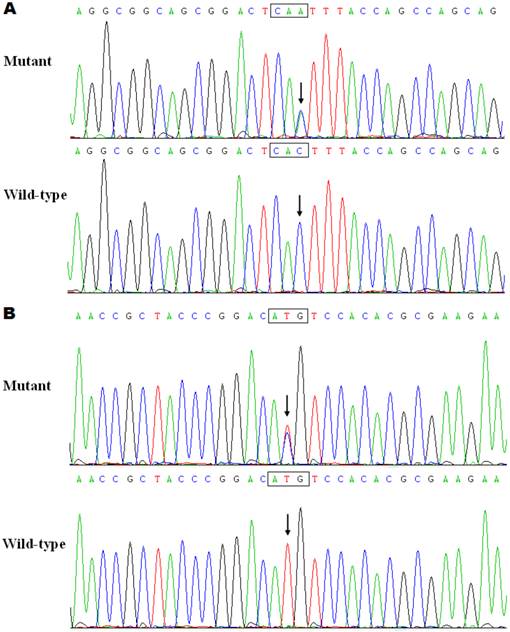
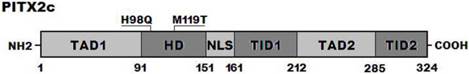
The exons and intron-exon boundaries of the PITX2c gene was sequenced in the 150 unrelated patients with CHD, and 2 heterozygous sequence variations in PITX2c were identified in 2 out of 150 patients, respectively, with a mutational prevalence of about 1.33% based on the patient population. Specifically, a substitution of adenine for cytosine at the third nucleotide of codon 98 of the PITX2c gene (c.294C>A), equivalent to the transition of histidine into glutamine at amino acid position 98 (p.H98Q), was identified in a male patient with atrial septal defect who was 12 years old. A transversion of thymine into cytosine in the second nucleotide of codon 119 (c.356T>C), predicting the change of methionine into threonine at amino acid 119 (p.M119T), was identified in a 3-year-old female patient with atrial septal defect. The mutation carriers had no apparent malformations in the eyes, teeth, or umbilicus, and had no positive family history. No relatives from the mutation carriers were available for genotyping PITX2c. The sequence electropherograms showing the identified heterozygous PITX2c variations in contrast to corresponding control sequences are illustrated in Figure 1. A schematic diagram of PITX2c showing the structural domains and the locations of the detected mutations is presented in Figure 2 [35,36]. The variations were neither observed in 400 control chromosomes nor reported in the EVS's and NCBI's SNP databases, which were consulted again on May 31, 2013.
Sequence electropherograms showing the PITX2c mutations in contrast to the corresponding controls. The arrow indicates the heterozygous nucleotides of C/A in one patient or T/C in another patient (mutant); or the homozygous nucleotides of C/C or T/T in the corresponding control individuals (wild-type). The rectangle denotes the nucleotides comprising a codon of PITX2c.
Schematic diagram of PITX2c protein structure with the atrial septal defect related mutations shown. The mutations identified in patients with atrial septal defects are shown above the structural domains. NH2 means amino-terminus; TAD1, transcriptional activation domain 1 (amino acids 1-90); HD, homeodomain (amino acids 91-151); NLS, nuclear localization signal (amino acids 145-161); TID1, transcriptional inhibitory domain 1 (amino acids 162-212); TAD2, transcriptional activation domain 2 (amino acids 213-285); TID2, transcriptional inhibitory domain 2 (amino acids 286-324); and COOH, carboxyl-terminus.
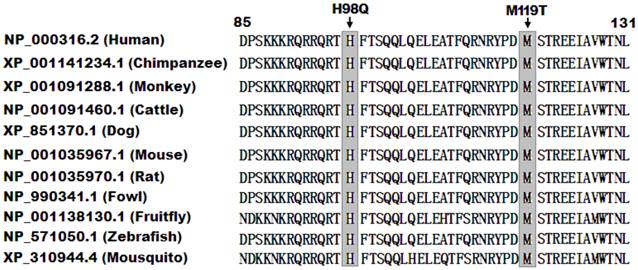
Alignment of multiple PITX2c protein sequences
A cross-species alignment of multiple PITX2c protein sequences displayed that the affected amino acids were completely conserved evolutionarily (Figure 3), indicating that the amino acids are functionally important.
Alignment of multiple PITX2c protein sequences across species. The altered amino acids of p.H98 and p.M119 are completely conserved evolutionarily.
Causative potential of PITX2c sequence variations
The PITX2c sequence variations of c.294C>A and c.356T>C were both automatically predicted to be disease-causing, with the same P value of 1.000. No SNPs in the altered regions were found in MutationTaster database. These PITX2c sequence variations were also predicted by PolyPhen-2 to be probably damaging, with the same score of 1.000 (sensitivity 0.00; specificity 1.00).
Transcriptional activity of the PITX2c mutants
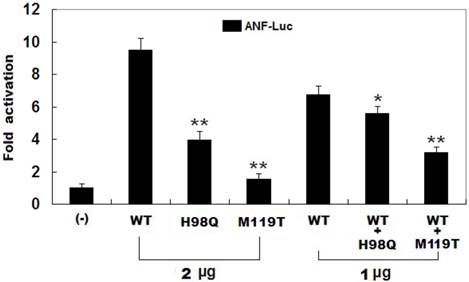
As shown in Figure 4, the same amounts of wild-type PITX2c (2 μg), H98Q-mutant PITX2c (2 μg) and M119T-mutant PITX2c (2 μg) activated the ANF promoter by ~10-fold, ~4-fold and ~2-fold increase, respectively, when compared with the empty plasmid. When the same amount of wild-type PITX2c (1 μg) was transfected in combination with H98Q-mutant PITX2c (1 μg) or M119-mutant PITX2c (1 μg), the induced activation of the ANF promoter was~6-fold or ~3-fold increase compared with the empty plasmid. These data indicate that both PITX2c mutants are associated with significantly reduced activation activity compared with their wild-type counterpart.
Functional defects resulted from PITX2c mutations. Activation of atrial natriuretic factor (ANF) promoter driven luciferase reporter in CHO cells by PITX2c wild-type PITX2c (WT) or mutant PITX2c (H98Q or M119T), alone or in combination, demonstrated significantly reduced transactivational activity caused by mutant proteins. Experiments were performed in triplicate, and mean and standard deviations are shown. ** indicates p < 0.001 and * denotes p < 0.005, when compared with the same amount (2 μg) of wild-type PITX2c.
Discussion
In the present study, two novel heterozygous mutations of PITX2c, p.H98Q and p.M119, were identified in 2 unrelated patients with atrial septal defects. The mutant alleles were absent in the 400 control chromosomes from an ethnically matched control population. A cross-species alignment of PITX2c protein sequences showed that the altered amino acids were completely conserved evolutionarily. The p.H98Q and p.M119 variants were both predicted to be pathogenic mutations by MutationTaster and PolyPhen-2, and the biochemical analysis revealed that the PITX2c mutant proteins were consistently associated with significantly reduced transcriptional activity. Therefore, it is likely that functionally impaired PITX2c is responsible for CHD in these patients. To the best knowledge of the authors, this is the first report on the association of PITX2c loss-of-function mutations with enhanced susceptibility to CHD in humans.
PITX2 is a member of the bicoid-like homeodomain transcription factor family. To date, 4 different PITX2 transcripts, derived from differential mRNA splicing and alternative promoter usage, have been discovered, of which PITX2a, PITX2b, and PITX2c differ only in their amino-termini and have been identified in human, mice, chick, zebrafish, and xenopus, while the fourth isoform, PITX2d, which lacks the amino-terminal domain and most of the homeodomain, has only been observed in humans. The unique amino-termini of PITX2a, PITX2b, and PITX2c modify their ability to activate transcription in a cell-type and promoter-dependent manner. The homeodomain is responsible for recognizing specific DNA sequences 5′-TAATCC-3′, which is essential for DNA binding, nuclear translocation, and interaction with other transcription factors [37]. Human PITX2c gene maps to chromosome 4q25, which consists of three exons coding for a protein of 324 amino acids [38]. PITX2c is expressed asymmetrically in the developing and adult heart, playing a crucial role in normal cardiovascular genesis and maturation [39]. The PITX2c mutations of p.H98Q and p.M119 identified in present study is located in homeodomain, thus may be expected to exert influence on the transcriptional activity of PITX2c by interfering with their DNA binding or subcellular trafficking.
It has been substantiated that PITX2c is an upstream regulator of multiple target genes expressed in the heart during embryogenesis, including the gene that encodes ANF [40]. Therefore, the functional effect of a PITX2c mutation can be explored by assay of the transcriptional activity of the ANF promoter in cells expressing PITX2c mutant in contrast to its wild-type counterpart. In this study, the functional characteristics of 2 novel PITX2c mutations identified in CHD patients were investigated by transcriptional activity analysis and the results showed that the mutations was both associated with a significantly reduced transcriptional activity on a downstream gene ANF, suggesting that dysfunctional PITX2c resulted from mutations is potentially an alternative pathological mechanism of CHD.
That functionally impaired PITX2c predisposes to congenital cardiovascular defects has been demonstrated in animal models. In mice, PITX2c was expressed predominantly in trabecular and septal myocardium with a strong expression bias in myocardium associated with endocardial cushions of the atrioventricular canal and outflow tract, which are essential for cardiac septation [41], and targeted deletion of the PITX2c gene led to embryonic death attributed to cardiovascular defects, including double-outlet right ventricle, atrial isomerism, atrial septal defect, ventricular septal defect, and abnormal aortic arch [32]. In Xenopus, knockdown of PITX2c by RNA interference gave rise to defects in the cardiac outflow tract and atrial septation [23]. These experimental findings corroborate an exquisite sensitivity of the developing cardiovascular system to the level of PITX2c.
Previous studies have unveiled multiple downstream genes transactivated by PITX2c [28], and loss-of-function mutations in several target molecules have been causally implicated in CHD, including NKX2.5, GATA4, CX40, and CX43 [18,19,42-46]. Therefore, mutated PITX2c may confer susceptibility to CHD by interfering with the expressions of such cardiac-spesific target genes.
In conclusion, this work firstly associates functionally compromised PITX2c with increased vulnerability to CHD in humans, providing novel insight into the molecular mechanism of CHD and suggesting potential implications for the early prophylaxis and allele-specific treatment of CHD.
Acknowledgements
The authors are really grateful to the participants for their dedication to the study. This work was supported in part by grants from the National Natural Science Fundation of China (81070153, 81270161 and 81271927), the Personnel Development Foundation of Shanghai, China (2010019), and the Natural Science Fund of Shanghai, China (10ZR1428000).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB; American Heart Association Statistics Committee, Stroke Statistics Subcommittee. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2-220
2. Warnes CA, Williams RG, Bashore TM, Child JS, Connolly HM, Dearani JA, del Nido P, Fasules JW, Graham TP Jr, Hijazi ZM, Hunt SA, King ME, Landzberg MJ, Miner PD, Radford MJ, Walsh EP, Webb GD. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to develop guidelines on the management of adults with congenital heart disease). Circulation. 2008;118:e714-833
3. Buys R, Cornelissen V, Van De Bruaene A, Stevens A, Coeckelberghs E, Onkelinx S, Thomaes T, Delecluse C, Budts W, Vanhees L. Measures of exercise capacity in adults with congenital heart disease. Int J Cardiol. 2011;153:26-30
4. Müller J, Hess J, Hager A. Exercise performance and quality of life is more impaired in Eisenmenger syndrome than in complex cyanotic congenital heart disease with pulmonary stenosis. Int J Cardiol. 2011;150:177-81
5. Teixeira FM, Coelho RM, Proença C, Silva AM, Vieira D, Vaz C, Moura C, Viana V, Areias JC, Areias ME. Quality of life experienced by adolescents and young adults with congenital heart disease. Pediatr Cardiol. 2011;32:1132-8
6. Müller J, Hess J, Hager A. Minor symptoms of depression in patients with congenital heart disease have a larger impact on quality of life than limited exercise capacity. Int J Cardiol. 2012;154:265-9
7. Shedeed SA, Elfaytouri E. Brain maturity and brain injury in newborns with cyanotic congenital heart disease. Pediatr Cardiol. 2011;32:47-54
8. Perry JC. Sudden cardiac death and malignant arrhythmias: the scope of the problem in adult congenital heart patients. Pediatr Cardiol. 2012;33:484-90
9. Silka MJ, Bar-Cohen Y. A contemporary assessment of the risk for sudden cardiac death in patients with congenital heart disease. Pediatr Cardiol. 2012;33:452-60
10. Knirsch W, Nadal D. Infective endocarditis in congenital heart disease. Eur J Pediatr. 2011;170:1111-27
11. Murakami T, Niwa K, Yoshinaga M, Nakazawa M. Factors associated with surgery for active endocarditis in congenital heart disease. Int J Cardiol. 2012;157:59-62
12. Cheng HH, Almodovar MC, Laussen PC, Wypij D, Polito A, Brown DW, Emani SM, Pigula FA, Allan CK, Costello JM. Outcomes and risk factors for mortality in premature neonates with critical congenital heart disease. Pediatr Cardiol. 2011;32:1139-46
13. Verheugt CL, Uiterwaal CS, van der Velde ET, Meijboom FJ, Pieper PG, Sieswerda GT, Plokker HW, Grobbee DE, Mulder BJ. The emerging burden of hospital admissions of adults with congenital heart disease. Heart. 2010;96:872-8
14. Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943-8
15. Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, Elixson M, Warnes CA, Webb CL; American Heart Association Council on Cardiovascular Disease in the Young. Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:2995-3014
16. Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL; American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015-38
17. Cecchetto A, Rampazzo A, Angelini A, Bianco LD, Padalino M, Stellin G, Daliento L. From molecular mechanisms of cardiac development to genetic substrate of congenital heart diseases. Future Cardiol. 2010;6:373-93
18. Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281:108-11
19. Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443-7
20. Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li RG, Xu YJ, Yang YQ. GATA5 loss-of-function mutations underlie tetralogy of fallot. Int J Med Sci. 2013;10:34-42
21. Zheng GF, Wei D, Zhao H, Zhou N, Yang YQ, Liu XY. A novel GATA6 mutation associated with congenital ventricular septal defect. Int J Mol Med. 2012;29:1065-71
22. Liu C, Liu W, Palie J, Lu MF, Brown NA, Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002;129:5081-91
23. Dagle JM, Sabel JL, Littig JL, Sutherland LB, Kolker SJ, Weeks DL. Pitx2c attenuation results in cardiac defects and abnormalities of intestinal orientation in developing Xenopus laevis. Dev Biol. 2003;262:268-81
24. Bamforth SD, Bragança J, Farthing CR, Schneider JE, Broadbent C, Michell AC, Clarke K, Neubauer S, Norris D, Brown NA, Anderson RH, Bhattacharya S. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat Genet. 2004;36:1189-96
25. Mommersteeg MT, Brown NA, Prall OW, de Gier-de Vries C, Harvey RP, Moorman AF, Christoffels VM. Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ Res. 2007;101:902-9
26. Galli D, Domínguez JN, Zaffran S, Munk A, Brown NA, Buckingham ME. Atrial myocardium derives from the posterior region of the second heart field, which acquires left-right identity as Pitx2c is expressed. Development. 2008;135:1157-67
27. Lozano-Velasco E, Chinchilla A, Martínez-Fernández S, Hernández-Torres F, Navarro F, Lyons GE, Franco D, Aránega AE. Pitx2c modulates cardiac-specific transcription factors networks in differentiating cardiomyocytes from murine embryonic stem cells. Cells Tissues Organs. 2011;194:349-62
28. Clauss S, Kääb S. Is Pitx2 growing up? Circ Cardiovasc Genet. 2011;4:105-7
29. Douglas YL, Jongbloed MR, Deruiter MC, Gittenberger-de Groot AC. Normal and abnormal development of pulmonary veins: state of the art and correlation with clinical entities. Int J Cardiol. 2011;147:13-24
30. Lin CR, Kioussi C, O'Connell S, Briata P, Szeto D, Liu F, Izpisúa-Belmonte JC, Rosenfeld MG. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature. 1999;401:279-82
31. Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643-51
32. Liu C, Liu W, Lu MF, Brown NA, Martin JF. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development. 2001;128:2039-48
33. Yang YQ, Xu YJ, Li RG, Qu XK, Fang WY, Liu X. Prevalence and spectrum of PITX2c mutations associated with familial atrial fibrillation. Int J Cardiol. 2013 [Epub ahead of print]
34. Strungaru MH, Footz T, Liu Y, Berry FB, Belleau P, Semina EV, Raymond V, Walter MA. PITX2 is involved in stress response in cultured human trabecular meshwork cells through regulation of SLC13A3. Invest Ophthalmol Vis Sci. 2011;52:7625-33
35. Footz T, Idrees F, Acharya M, Kozlowski K, Walter MA. Analysis of mutations of the PITX2 transcription factor found in patients with Axenfeld-Rieger syndrome. Invest Ophthalmol Vis Sci. 2009;50:2599-606
36. Acharya M, Lingenfelter DJ, Huang L, Gage PJ, Walter MA. Human PRKC apoptosis WT1 regulator is a novel PITX2-interacting protein that regulates PITX2 transcriptional activity in ocular cells. J Biol Chem. 2009;284:34829-38
37. Simard A, Di Giorgio L, Amen M, Westwood A, Amendt BA, Ryan AK. The Pitx2c N-terminal domain is a critical interaction domain required for asymmetric morphogenesis. Dev Dyn. 2009;238:2459-70
38. Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392-9
39. Kirchhof P, Kahr PC, Kaese S, Piccini I, Vokshi I, Scheld HH, Rotering H, Fortmueller L, Laakmann S, Verheule S, Schotten U, Fabritz L, Brown NA. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet. 2011;4:123-33
40. Ganga M, Espinoza HM, Cox CJ, Morton L, Hjalt TA, Lee Y, Amendt BA. PITX2 isoform-specific regulation of atrial natriuretic factor expression: synergism and repression with Nkx2.5. J Biol Chem. 2003;278:22437-45
41. Furtado MB, Biben C, Shiratori H, Hamada H, Harvey RP. Characterization of Pitx2c expression in the mouse heart using a reporter transgene. Dev Dyn. 2011;240:195-203
42. Britz-Cunningham S, Shah M, Zuppan C, Fletcher W. Mutations of the connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N Engl J Med. 1995;332:1323-9
43. Dasgupta C, Martinez A, Zuppan C, Shah M, Bailey L, Fletcher W. Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (dgge). Mutat Res. 2001;479:173-86
44. Wang B, Wen Q, Xie X, Liu S, Liu M, Tao Y, Li Z, Suo P, Shen A, Wang J, Ma X. Mutation analysis of Connexon43 gene in Chinese patients with congenital heart defects. Int J Cardiol. 2010;145:487-9
45. Krüger O, Maxeiner S, Kim JS, van Rijen HV, de Bakker JM, Eckardt D, Tiemann K, Lewalter T, Ghanem A, Lüderitz B, Willecke K. Cardiac morphogenetic defects and conduction abnormalities in mice homozygously deficient for connexin40 and heterozygously deficient for connexin45. J Mol Cell Cardiol. 2006;41:787-97
46. Soemedi R, Topf A, Wilson IJ, Darlay R, Rahman T, Glen E, Hall D, Huang N, Bentham J, Bhattacharya S, Cosgrove C, Brook JD, Granados-Riveron J, Setchfield K, Bu'lock F, Thornborough C, Devriendt K, Breckpot J, Hofbeck M, Lathrop M, Rauch A, Blue GM, Winlaw DS, Hurles M, Santibanez-Koref M, Cordell HJ, Goodship JA, Keavney BD. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum Mol Genet. 2012;21:1513-20
Author contact
![]() Corresponding author: Wei-Yi Fang, Tel: +86-21-56051080; Fax: +86-21-66371663; Email: xkfangwycom. Yi-Qing Yang, Tel: +86-21-62821990; Fax: +86-21-62821105; Email: dryyqedu.cn
Corresponding author: Wei-Yi Fang, Tel: +86-21-56051080; Fax: +86-21-66371663; Email: xkfangwycom. Yi-Qing Yang, Tel: +86-21-62821990; Fax: +86-21-62821105; Email: dryyqedu.cn