Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
An overview of HIF-1
The mutual relationship between...
Hyperglycemia and impairment of...
The negative effects of...
The role of reactive oxygen...
Increased sensitivity to VHL...
Participation of osmolarity and...
Other possible mechanisms
A summary of the above mechanisms
Adverse aspects: activation of...
Possible therapies
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(10):1412-1421. doi:10.7150/ijms.5630 This issue Cite
Review
The Possible Mechanisms Underlying the Impairment of HIF-1α Pathway Signaling in Hyperglycemia and the Beneficial Effects of Certain Therapies
Haijuan Xiao1, Zhenyang Gu2, Guoxing Wang3, Tongfeng Zhao1 ![]()
1. Department of Endocrinology, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou 510655, China.
2. Department of Hematology, Division of internal medicine, Chinese PLA General Hospital, Beijing 100853, China.
3. Department of Endocrinology, The Affiliated Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou 310020, China.
Received 2012-11-30; Accepted 2013-7-23; Published 2013-8-22
Abstract
Hypoxia-inducible factor 1 alpha (HIF-1α), an essential transcription factor which mediates the adaptation of cells to low oxygen tensions, is regulated precisely by hypoxia and hyperglycemia, which are major determinants of the chronic complications associated with diabetes. The process of HIF-1α stabilization by hypoxia is clear; however, the mechanisms underlying the potential deleterious effect of hyperglycemia on HIF-1α are still controversial, despite reports of a variety of studies demonstrating the existence of this phenomenon. In fact, HIF-1α and glucose can sometimes influence each other: HIF-1α induces the expression of glycolytic enzymes and glucose metabolism affects HIF-1α accumulation in some cells. Although hyperglycemia upregulates HIF-1α signaling in some specific cell types, we emphasize the inhibition of HIF-1α by high glucose in this review. With regard to the mechanisms of HIF-1α impairment, the role of methylglyoxal in impairment of HIF-1α stabilization and transactivation ability and the negative effect of reactive oxygen species (ROS) on HIF-1α are discussed. Other explanations for the inhibition of HIF-1α by high glucose exist: the increased sensitivity of HIF-1α to Von Hippel-Lindau (VHL) machinery, the role of osmolarity and proteasome activity, and the participation of several molecules. This review aims to summarize several important developments regarding these mechanisms and to discuss potentially effective therapeutic techniques (antioxidants eicosapentaenoic acid (EPA) and metallothioneins (MTs), pharmaceuticals cobalt chloride (CoCl2), dimethyloxalylglycine (DMOG), desferrioxamine (DFO) and gene transfer of constitutively active forms of HIF-1α) and their mechanisms of action for intervention in the chronic complications in diabetes.
Keywords: Hypoxia-inducible factor 1 alpha (HIF-1α), Hyperglycemia, Prolyl hydroxylases (PHDs), Methylglyoxal (MGO), Reactive oxidative species (ROS), Therapy.
An overview of HIF-1
Hypoxia-inducible factor 1 (HIF-1), which is a heterodimeric transcription factor composed of two subunits: HIF-1α and HIF-1β, regulates a number of essential pathways in the adaptive responses of cells to hypoxic conditions [1]. These two subunits are both basic helix-loop-helix (bHLH) proteins of the PAS family (PER, AHR, ARNT and SIM family); however, they display different responses to O2 concentrations: HIF-1β is a non-oxygen-responsive nuclear protein and is constitutively expressed; while the levels and activity of HIF-1α are tightly regulated by cellular O2 concentrations [1, 2]. Under normoxia, HIF-1α has an extremely short half-life of less than five minutes, being continuously synthesized and degraded [3]. The degradation of HIF-1α is mediated by the hydroxylation of two prolyl residues (402 and 564) in the oxygen-dependent degradation domain (ODD) by the specific prolyl hydroxylases (PHDs) [4], which require oxygen and 2-oxoglutarate, as co-substrates, and iron (Fe2+) and ascorbate, as co-factors [5, 6]. Prolyl hydroxylation of HIF-1α is required for binding of the von Hippel-Lindau protein (VHL), which forms part of the E3 ligase complex (a ubiquitin ligase complex) that targets HIF-1α for polyubiquitination and subsequent proteasomal degradation [7]. Besides this canonical pathway, there is evidence to demonstrate that the degradation of HIF-1α may also occur in oxygen- or VHL- independent ways [8, 9]. While under hypoxic conditions, the hydroxylation of prolyl residues is inhibited, thus HIF-1α evades VHL-mediated proteasomal destruction leading to accumulation.
Apart from HIF-1α protein stability, its transactivation ability also deserves extensive attention. This function is related to two essential domains: the amino-terminal transactivation domain (NTAD) and the carboxy-terminal transactivation domain (CTAD), which is the binding location of the transcriptional co-activators p300/CREB binding protein (CBP) with HIF-1α [10, 11]. The NTAD overlaps with the ODD; therefore, NTAD transcriptional activity and HIF-1α protein stability are coupled. In contrast to the NTAD, regulation of the CTAD activity is connected with the hydroxylation of a key asparagine residue (Asn-803) through a reaction catalyzed by factor-inhibiting HIF-1 (FIH-1) (another iron- and oxoglutarate- dependent oxygenase), which impairs the association of CTAD and p300/CBP [10]. Therefore, hydroxylation depending on O2 offers a direct mechanism by which changes in the cellular O2 concentration can be transduced to the nucleus as changes in the half-life and transactivation function of HIF-1α [12].
Once the HIF-1α pathway is activated, under hypoxic conditions for example, the hydroxylation of proline residues in the ODD ceases; consequently, HIF-1α escapes proteasomal degradation and accumulates and is translocated to nucleus, where it dimerizes with HIF-1β and binds to target genes at hypoxia response elements (HREs) [13, 14]. At the same time, the hydroxylation of the key asparagine residue in the CTAD is also inhibited, and p300/CBP interacts with the CTAD [14, 15]. In fact, the binding of the cysteine/histidine-rich (CH1) region of the coactivator p300 to HIF-1α CTAD is essential for HIF-1 transcriptional activity [15]. HREs, which contain the core pentanucleotide sequence 5'-(A/G)CGTG-3' and are often found in the promoter or enhancer sequences of HIF-1α target genes, are defined by their function as cis-acting elements sufficient for the mediation of a transcriptional response to hypoxia after the binding of HIF-1α [13, 16].
It has been demonstrated that hundreds of genes are HIF-1α regulated and over 90 genes are direct HIF-1α targets through the functional demonstration of a HRE [17, 18]. Furthermore, these genes have been shown to be involved in a plethora of adaptation and survival mechanisms, such as angiogenesis, wound healing, anaerobic glucose energy metabolism, erythropoiesis, and cell growth, proliferation, differentiation, survival and apoptosis [19]. Among these, several well-known genes include vascular endothelial growth factor (VEGF), haem oxygenase 1 (HO-1), nitric oxide synthase (NOS), endothelin, erythropoietin (EPO), lactate dehydrogenase A (LDH-A), Glucose transporter 1 (GLUT-1), Glucose transporter 3 (GLUT-3), C-X-C chemokine receptor type 4 (CXCR4) and the CXCR 4-ligand stromal cell -derived factor-1 (SDF-1) and p53 [16, 19, 20-25].
HIF-1β, also known as the aryl hydrocarbon receptor nuclear translocator (ARNT), plays an important role in maintaining glucose-stimulated insulin secretion (GSIS) from pancreatic β cells [26]. Elevated glucose concentrations can result in a repression of HIF-1β, and this is consistent with the observation that the levels of HIF-1β were reduced in islets and livers obtained from patients with type 2 diabetes. This contributed to the impaired secretion function of beta-cells and increased hepatic gluconeogenesis, increased lipogenic gene expression, and low serum beta-hydroxybutyrate in human type 2 diabetes [27]. There has been a report that HIF-1α is vital for the expression of HIF-1β and beta-cell function and reserve, indicating a functional as well as a structural interaction between these two subunits [28].
The mutual relationship between glucose and HIF-1α
Apart from hypoxia, glucose also affects the expression and activation of HIF-1α in human pharyngeal carcinoma and fibrosarcoma cells [29, 30]. In fact, the relationship between glucose and HIF-1α is sometimes mutual. On the one hand, it has been shown that HIF-1α regulates the expression of nearly all the enzymes involved in the process of glycolysis and of GLUT1 and GLUT3 which mediate cellular glucose uptake [31]. On the other hand, glucose, glucose uptake and glycolysis influence the stability and activation of HIF-1α in human pharyngeal carcinoma and fibrosarcoma cells and rat cardiac myocytes [25, 29, 30].
Although glucose alone in the absence of hypoxia is not enough to activate the HIF-1α pathway, normal glucose concentrations are critical for HIF-1α protein expression and activation in response to hypoxia [29]. The reduction of the glucose concentration form 5.5 to 0.55 mM almost completely abolished hypoxic HIF-1α accumulation in FaDu human pharyngeal carcinoma and HT 1080 human fibrosarcoma cells [29]. Glucose metabolism also affects the expression of HIF-1α. The inhibition of glycolysis reduced hypoxic HIF-1α protein accumulation in HT1080 cells, which happened on a translational level but was independent of the activation of PHD [30]. It should be noted that these two studies showing the importance of glucose and glucose metabolism to HIF-1α were both tumor-related.
However, adverse conclusions have also been reported. Malhotra et al. demonstrated that glucose, glucose uptake and glycolysis as well as GLUT1 overexpression could promote the ubiquitination of HIF-1α in hypoxic rat cardiac myocytes and thereby enhanced its degradation by the ubiquitin proteasomal pathway [25]. They indicated that this prohibitive effect of glucose and glucose metabolism on HIF-1α stability served as a feedback mechanism, whereby HIF-1α accelerated the expression and activation of GLUT1 and induced glucose uptake and glycolysis which in turn induced HIF-1α degradation [25].
Hyperglycemia and impairment of the HIF-1α pathway
There have been a mass of independent studies of the reduced levels of HIF-1α in the cells or tissues obtained from patients with diabetes or from animal models of diabetes, and in cells cultured in high glucose medium, which lead to a consensus that hyperglycemia is responsible for compromised HIF-1α protein levels and transactivation function. Although the detailed molecular mechanisms underlying impairment of HIF-1α in diabetes or in high glucose remain poorly understood, some recent studies envision this inhibition pathway.
The negative effects of methylglyoxal
Methylglyoxal (MGO), a highly reactive α-oxoaldehyde and dicarbonyl mainly formed as a by-product of glycolysis, is increased in the setting of high glucose-induced oxidative stress and forms stable adducts primarily with arginine residues of intracellular proteins [32]. In detail, hyperglycemia-induced superoxide formation leads to inhibition of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and subsequent upstream triose phosphate glycolytic metabolite accumulation, which produces more MGO [33]. Overexpression of glyoxalase I (GLO1), the rate-limiting enzyme of MGO catabolism, reverses some of the negative effects of MGO on HIF-1α [34, 35]. The accumulation of MGO in elevated glucose concentrations generates an inhibition of HIF-1α stability and transactivation ability through three distinct ways.
Ceradini et al. showed that high glucose-induced MGO led to the covalent modification of HIF-1α at arginine 17 (Arg-17) and arginine 23 (Arg-23) of the bHLH domain (the locus mediating the interaction of HIF-1α and HIF-1β), which decreased its heterodimer formation with HIF-1β and further inhibited HIF-1 binding to HRE of its target genes (Fig. 1A) [34]. The impairment of HIF-1α by MGO resulted not only in reduced transcription of SDF-1 (the endothelial progenitor cells (EPCs) mobilizing chemokines) and VEGF (a growth factor regulating growth and differentiation of recruited EPCs) in hypoxic mouse fibroblasts, but also in decreased transcription of SDF-1 receptor CXCR4 and eNOS (an enzyme essential for EPC mobilization) in hypoxic EPCs, which led to defective ischemia-induced vasculogenesis in diabetic mice [34].
Possible mechanisms underlying the impairment of the HIF-1α pathway by hyperglycemia (1). A, B, C, D: the effects of MGO on HIF-1α; A: The covalent modification of HIF-1α leads to decreased dimerization of HIF-1α and HIF-1β, and further reduces the binding of HIF-1 and HRE; B: The covalent modification of p300 leads to inhibition of the interaction of CTAD and p300, which decreases the transactivation ability of HIF-1α; C: The covalent modification of HIF-1α increases its association with HSP40/70, which recruits CHIP and leads to HIF-1α proteasomal degradation; D: The exposure of RPE cells to MGO leads to destabilization of HIF-1α; E: Hyperglycemia increases the sensitivity of hydroxyl HIF-1α to VHL machinery; F: Hyperglycemia suppresses CTAD and NTAD, and finally reduces the transactivation ability of HIF-1α; G: In HDF and HDMEC, osmolarity is one of the mechanisms of hyperglycemia action; H: The impairment of HIF-1α stabilization by hyperglycemia is mediated by proteasomal degradation. CBP: CREB binding protein; CHIP: carboxyl terminus of the Hsc70-interacting protein; CREB: cAMP-response element binding protein; CTAD: carboxy-terminal transactivation domain; HDF: human dermal fibroblast; HDMEC: human dermal microvascular endothelial cells; HIF-1: hypoxia-inducible factor 1; HREs: hypoxia response elements; Hsc70: heat-shock cognate protein 70; Hsp40/70: heat shock protein 40/70; MGO: methylglyoxal; NTAD: amino-terminal transactivation domain; OH: hydroxy; PHDs: prolyl hydroxylases; RPE: retinal pigment epithelial cells; Ub: ubiquitin; VHL: Von Hippel-Lindau protein.
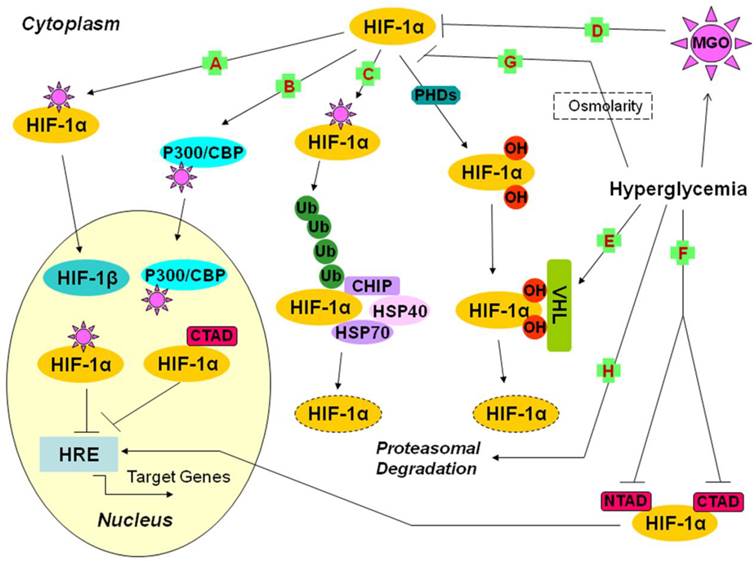
Furthermore, Thangarajah et al. indicated that MGO formed covalent interaction with p300, which prevented its binding to CTAD, and it was this decreased interaction of CTAD and p300 as a result of hyperglycemia that was responsible for the impaired transcriptional activation function of HIF-1α (Fig. 1B) [36, 37]. Experimental results demonstrating that the impairment of HIF-1α transactivation was maintained even when constitutive HIF-1α protein was overexpressed and when CTAD was unaffected by high glucose exposure supported this conclusion. The mutation of arginine 354 (Arg-354) of p300 prevented the modification of p300 and rescued its interaction with HIF-1α [36, 37]. High glucose-induced decreases in transactivation of HIF-1 led to impaired VEGF production in response to hypoxia, which resulted in reduced neovascularization in cells obtained from diabetic patients and impaired wound healing in ischemic diabetic animals [36, 37].
Moreover, Bento et al. demonstrated that HIF-1α-modification by MGO caused its increasing association with the molecular chaperone heat shock protein 40/70 (Hsp40/70) which recruited the carboxyl terminus of the heat-shock cognate protein 70 (Hsc70)-interacting protein (CHIP), a ubiquitin ligase, and led to polyubiquitination and proteasomal degradation (Fig. 1C) [38]. This process, which was independent of the recruitment of pVHL and did not require the hydroxylation of prolines, led to a dramatic decrease in HIF-1α transcriptional activity and subsequent loss of the cell response to hypoxia under conditions of high glucose. They pointed out that silencing of endogenous CHIP could stabilize HIF-1 under hypoxia in the presence of high glucose-induced MGO [38].
Increased levels of MGO in hyperglycemia induce HIF-1α and p300 modifications, which is sufficient to disrupt the interaction between HIF-1α/HIF-1β and HIF-1α/p300 and to cause proteasomal degradation of HIF-1α mediated by CHIP [34, 37, 38]. It is likely that these three mechanisms are not inconsistent, considering that the reduced interaction of HIF-1α/p300 and HIF-1α/HIF-1β by MGO promotes the number of modified and monomeric HIF-1α available to be degraded through a CHIP-mediated pathway [38]. Besides these three pathways, Bento and colleagues showed that MGO increased the levels of Ang2 and decreased the levels of HIF-1α and VEGF secreted by retinal pigment epithelial (RPE) cells, thus inducing an imbalance in the VEGF/Ang 2 ratio, which activated apoptosis, decreased proliferation of retinal endothelial cells and might contribute to endothelial dysfunction in diabetic retinopathy (Fig. 1D) [35].
The role of reactive oxygen species (ROS)
As mentioned above, hyperglycemia augments oxidative stress and induces the overproduction of ROS [39], which modulates HIF-1α regulation. A study has demonstrated that ROS, especially superoxide (O2-) degrades HIF-1α at the post-transcriptional level by activating a proline hydroxylase in the presence of iron and by increasing ubiquitin-proteasome activity (Fig. 2C) [40]. Apart from this direct action and the effects of MGO already discussed, there are other ways in which ROS represses HIF-1α. Here, we describe two key molecules that take part in this influential process: nitric oxide (NO) and Rac1.
Possible mechanisms underlying the impairment of the HIF-1α pathway by hyperglycemia (2). A, B, C: the roles of hyperglycemia-induced ROS; A: The reaction between O2- and NO results in a decrease in steady-state NO concentration and thereby reduces NO-induced HIF-1α accumulation and activation; B: Rac1 contributes to HIF-1α expression and transactivation; ROS inhibits HIF-1 expression through repression of Rac1 expression; C: ROS degrade HIF-1α by activating a proline hydroxylase in the presence of iron and by increasing ubiquitin-proteasome activity; D: The inhibition of HIF-1α might be involved in the role of TNF-α, angiotensin II and the insulin pathway (insulin, IGF-1, IGF-2). HIF-1: hypoxia-inducible factor 1; IGF: insulin-like growth factor; NO: nitric oxide; O2-: superoxide; PHDs: prolyl hydroxylases; ROS: reactive oxygen species; TNF-α: tumor necrosis factor-α.
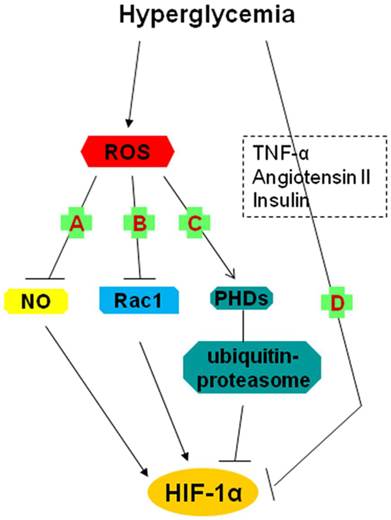
Bullock et al. reported that hyperglycemic brain ischemia in superoxide dismutase (SOD2-/+) mice significantly suppressed HIF-1α protein levels and exacerbated brain damage in the ischemic core region [41]. In detail, pre-ischemic hyperglycemia increased the production of O2- [42], which accumulated drastically because of the deletion of SODs which scavenged O2- [43]. Enhanced O2- levels reacted with NO, resulting in a decrease in steady-state NO concentrations and therefore altering the signaling response mediated by NO [44]. While NO induced the accumulation and activation of HIF-1α [45], diminished NO caused the inhibition of HIF-1α [44], this is, a decline in steady-state NO concentrations occurred when increased O2- in high glucose reacted with NO, which accounted for a proportional reduction in HIF-1α levels (Fig. 2A).
Rac1, a small GTPase of the Rho family, is considered as a key determinant of intracellular redox status and plays an essential role in the induction of HIF-1α protein expression and transcriptional activity in response to hypoxia [46]. Impairment in HIF-1α mRNA expression and in Rac1 mRNA expression associated with an increase in infarct size in ischemic hearts of diabetic rats and isolated ischemic rats under conditions of high glucose was reported by Marfella et al. [47]. These effects of high glucose could be prevented by glutathione, a powerful antioxidant, which suggested that prevailing oxidative stress played an essential role in mediating the response of HIF-1 to ischemia, with the help of Rac1 [47]. In other words, the improved ROS levels in hyperglycemia inhibited Rac1 expression and ultimately repressed HIF-1α expression (Fig. 2B).
Increased sensitivity to VHL machinery and the inhibition of CTAD and NTAD
Botusan et al. demonstrated that hyperglycemia destabilized HIF-1α in the primary fibroblasts and wounds of db-/db- diabetic mice, and this effect of high glucose was dependent on the increasing sensitivity to VHL machinery (Fig. 1E). The observation that hyperglycemia did not modulate HIF-1α in cells lacking functional pVHL confirmed this decrease depending on the VHL-mediated degradation mechanism [48]. What is more, NTAD and CTAD, which were critically significant for HIF-1α transactivation function, were also inhibited by high glucose (Fig. 1F) [48]. This article did not discuss the mechanism by which NTAD and CTAD were suppressed by high glucose; further studies were required to elucidate this issue. In summary, both the protein expression and transactivation activity of HIF-1α were impaired in high glucose environments, which brought about decreased expression of HIF-1 target genes and delayed wound healing in diabetes.
Participation of osmolarity and proteasome activity
The interference with the hypoxia-dependent stabilization and functional activity of HIF-1α against proteasomal degradation by high glucose in human dermal fibroblast (HDF) and human dermal microvascular endothelial cells (HDMEC), which led to a defective response of diabetic tissues to low oxygen tension, was shown in a study by Catrina and collaborators [54]. Lower levels of HIF-1 were detected in diabetic chronic foot ulcers compared with chronic venous ulcers. Detailed interference mechanisms were not described in this article, however, they demonstrated inhibition in a dose-dependent manner and confirmed the influence of two factors: osmolarity, in which the effect of hyperglycemia was mimicked by mannitol (Fig. 1G); proteasome activity, based on the observation that MG132 (carbobenzoxy-L-leucyl-L-leucyl-L-leucinal, a specific inhibitor of proteasomal activity) abolished the inhibitory effects of high glucose or mannitol on HIF-1α (Fig. 1H) [54].
Other possible mechanisms
Mace et al. indicated that HIF-1α protein levels and mRNA expression of its target genes (VEGF, HO-1 and NOS) were greatly decreased in diabetic cells (primary fibroblasts) and wounds and that sustained expression of HIF-1α following cobalt chloride (CoCl2) treatment or by gene transfer of a constitutively active form of HIF-1α (CMV-Hif-1α△ODD expression plasmid) restored the expression of HIF-1α and significantly accelerated wound healing in a diabetic mouse model [49]. The recovery of HIF-1α expression under high glucose exposure was sufficient to activate the downstream pathway, which was essential for the adaptive responses of cells to reduced oxygen availability and ischemia [11, 49]. As for the specific mechanisms underlying this impairment, further investigations were not described in this article although the possible role of three factors (Tumor necrosis factor-α (TNF-α), Angiotensin II (Ang II) and Insulin pathway (Insulin, Insulin-like growth factor 1 (IGF-1), Insulin-like growth factor 2 (IGF-2) ) in the reduction were mentioned (Fig. 2D) [50-53].
A summary of the above mechanisms
The studies above, conducted with precise experiments, described the effects of hyperglycemia on HIF-1α protein stability or its transactivation ability or both, or on HIF-1α mRNA expression. However, these reports do not constitute the whole story accounting for the inhibition of the HIF-1α pathway activation by high glucose; some other factors, such as advanced glycation end products (AGEs), NO, prostaglandin E2, might be involved in this downregulation process [55, 56, 57]. Furthermore, these studies are not fully consistent in some aspects. However, we should acknowledge the existence of two facts that might produce apparent discrepancies between these results: cell- and tissue-specific regulatory mechanisms encountered in different animal models and different cell culture conditions and systems [19]. It is likely that several, or all of these are the true causes of the negative effects of high glucose on HIF-1α pathway. Therefore, further experimentation combining animal models with cell culture systems are required to corroborate the conclusions we have gathered and to make novel discoveries.
Adverse aspects: activation of HIF-1α by high glucose
We have discussed several mechanisms explaining the impairment of HIF-1α by hyperglycemia in detail; however, there are also a number of studies demonstrating that HIF-1α is elevated and activated under conditions of high glucose through different pathways. Xiao et al. showed that high glucose concentrations accelerated HIF-1α protein synthesis and promoted its stability in cultured human retinal pigment epithelial cells [58]. Yan et al. indicated that high glucose upregulated HIF-1α protein levels and increased HIF-1α transcriptional activity in endothelial cells, which played an important role in high glucose-induced blood-brain barrier (BBB) permeability [59]. We explore more molecules involved in the positive effects of high glucose on HIF-1α in the following section.
It was reported that glucose stimulation of O2 consumption in rat pancreatic beta-cells induced intracellular hypoxia and activated HIF-1α, which played an important role in beta-cell adaptation to increased insulin demand under physiological conditions (physiological hypoxia) [60]. However, in diabetes, sustained activation of HIF-1α by hyperglycemia tended to reduce GSIS with consequent development of glucose intolerance [61]. Bensellam et al. suggested that high glucose resulted in a high metabolic demand and increased beta-cell O2 consumption; the following hypoxia and sustained HIF-1α activation contributed to the slow deterioration of beta-cell function [60].
There are three conserved E-box-like sequences in the HIF-1α gene promoters, and the carbohydrate response element (ChRE) is located in the proximal E-box-like sequences [62]. Carbohydrate response element binding protein (ChREBP), a glucose-responsive sensor, is a transcriptional factor binding to the ChRE; the binding of ChREBP to the HIF-1α promoter in glomerular mesangial cells exposed to high glucose mediates the upregulation of the HIF-1α mRNA by high glucose [63]. Furthermore, increased intracellular free calcium concentrations (Ca2+) in response to hyperglycemia activate CaM-dependent protein kinase II (CaMKII), which initiates a signaling cascade resulting in the phosphorylation of the cAMP response element binding protein (CREB); this phospho-activated CREB (p-CREB) induces target gene expression [64]. High glucose-induced HIF-1α expression in cultured rat retinal Müller cells depends on intracellular free Ca2+ and activation of the CaMKII- CREB pathway [64].
The effect of hyperglycemia on HIF-1 through the action of ROS is complicated: apart from the negative role that ROS induced by high glucose plays in HIF-1α levels [40, 41, 47], HIF-1α accumulation due to an inhibition of PHD activity by ROS has been reported by Köhl and colleagues [65]. This group postulated this concentration-dependent inhibition was associated with oxidation of the active site Fe2+, which was closely connected with cellular redox status [65]. What is more, Guo et al. demonstrated that high glucose promoted HIF-1α stabilization through regulation of the redox status in primary neurons exposed to hypoxia [66]. They showed that high glucose suppressed the generation of O2- and H2O2 and that ROS induced the degradation of HIF-1α in hypoxic neurons [66].
Marfella et al. observed an increase in basal HIF-1α mRNA expression in rat hearts, which suggested a pseudohypoxic state caused by hyperglycemia under non-hypoxic condition [47]. In spite of normal tissue PO2, the metabolic imbalances caused by hyperglycemia increased the cytosolic ratio of free NADH to NAD+. It was this increased ratio that led to pseudohypoxia [67]. The increased HIF-1α induced by hyperglycemic pseudohypoxia was associated with the role of NO [67], since hyperglycemic pseudohypoxia brought about an increased production of NO, and NO augmented HIF-1α accumulation [45].
We have discussed a number of mechanisms accounting for the effect of high glucose on the HIF-1α pathway; however, two questions remain to be answered: in a specific cell or tissue type under conditions of hyperglycemia, whether HIF-1α is impaired or enhanced and which mechanisms play a role in this process. To resolve these two questions, first, we have to acknowledge one premise: that the responses are various according to different types of cells and different conditions and second, more studies must be undertaken to explore the unknown scientific world. In this review, our emphasis is laid on the inhibition of HIF-1α pathway by high glucose; therefore, the therapies discussed below are targeted at the impairment of HIF-1α by hyperglycemia. We are dedicated to identifying measures to restore the expression and transactivation ability of HIF-1α under conditions of high glucose or diabetes.
Possible therapies
In fact, the ultimate significance of identifying the true mechanisms underlying the impairment of HIF-1α by high glucose is to find possible therapeutic approaches for the diseases associated with HIF-1α inhibition by hyperglycemia. Pharmacological intervention, gene transfer-based stabilization of HIF-1α and antioxidants such as eicosapentaenoic acid (EPA) and metallothioneins (MTs) are able to reverse HIF-1α defects in high glucose environments, and are likely to be of great clinical significance.
Local wound application of CoCl2 can correct the deficiencies in the levels of HIF-1α protein and VEGF mRNA and protein under conditions of hyperglycemia and increase HIF-1α DNA-binding activity. The effects of Co2+ appear to prevent prolyl and asparaginyl hydroxylases by exchanging with Fe2+ at the catalytic site [49, 68]. In addition, cobalt can replace the iron in heme [69], and there have also been studies demonstrating that Co2+ can be a substrate for ferrochelatase, the enzyme responsible for the incorporation of iron into protoporphyrin IX to make heme [70]. This causes the prevention of the oxygen signals and leads to chemical hypoxia [69]. There has been a study showing that cobalt reduces proteinuria and histological kidney injury in type 2 diabetic rats with nephropathy, which is attributed to the upregulation of HIF and HIF-regulated genes [71].
Desferrioxamine (DFO) and dimethyloxalylglycine (DMOG) both stabilize and activate HIF-1α by inhibiting hydroxylases via two mechanisms: competitive antagonism of α-ketoglutarate and iron chelators [72]. Both DMOG and DFO treatment improve angiogenesis and the healing process in diabetic animals [48]. However, in contrast to DMOG, DFO affects HIF-1α by scavenging hydroxyl radical-generating free iron generated by high-glucose induced oxidative stress. Through decreasing generation of ROS and preventing MGO modification of p300, DFO improves the HIF-1α/p300 interaction, augments HIF-1 transactivation and VEGF expression in response to high glucose exposure and thus enhances wound and ulcer healing and new vessel formation in diabetic ischemic tissues [37, 73].
In addition to the pharmacological usage mentioned above, recent studies have also begun to assess the therapeutic potential of gene therapy in diabetic animal models. Kajiwara et al. demonstrated that the usage of adenoviral vectors expressing a constitutively active HIF-1α hybrid (Ad2/HIF-1α/VP16) increased collateral development in a diabetic rat model [36]. Delivery of AdCA5 (an adenovirus encoding a constitutively active form of HIF-1α) was effective in blocking hyperglycemia-reduced VEGF protein expression and ameliorating maternal diabetes-induced embryonic vasculopathy [74]. In the leptin receptor deficient mouse (Leprdb-/-) model of type II diabetes, HIF-1α overexpression through introduction of a CMV-Hif-1α△ODD expression plasmid restored HIF-1α stabilization and significantly accelerates tissue repair [49].
Furthermore, since ROS leads to the impairment of HIF-1α pathway, the application of some kind of antioxidant (EPA and MT) could also ameliorate diseases related to HIF-1α defects. EPA, an n-3 polyunsaturated fatty acid abundant in fish oil, upregulates local HIF-1α expression and augments the HIF-1α response in diabetic kidney disease by suppressing ROS generation and mitochondrial apoptosis and thereby ameliorating hyperglycemia-induced renal tubular injury and dysfunction [75]. MT is a small protein, with high cysteine content that protects cells or tissues from diabetic-induced oxidative damage due to its powerful antioxidant defense against ROS and/or reactive nitrogen species (RNS) [76]. Feng et al. showed that MT relieved the high glucose suppression of HIF-1α activity and rescued HIF-1α transcriptional activity in cardiomyocytes under diabetic conditions [77]. Cai et al. concluded that MT could attenuate cardiac cell death and prevent diabetic cardiomyopathy [76].
The overexpression of SOD or GLO1 as a therapy is also worthy of consideration. SOD scavenges ROS (especially O2-) and GLO1 increases detoxification of MGO, thereby enhancing the stability and function of HIF-1α [19, 34]. It has been reported that GLO1 expression can prevent MGO-induced impairment of the HIF-1α pathway in response to hypoxia and high glucose levels and thus, enhance neoangiogenesis and wound healing in diabetes [19, 34, 35, 37, 38]. However, it should be noted that GLO1 overexpression in high glucose environments requires a concomitant increase in glutathione (GSH) levels in cells, because GLO1 is a GSH-dependent enzyme and GSH is defective in diabetic tissues [78, 79]. Otherwise, it is likely that GLO1 function is compromised in diabetes.Perspectives
This review summarizes recent studies of the normal physiological regulation of HIF-1α, the mutual relationship between glucose and HIF-1α and the mechanisms underlying the impairment and enhancement of HIF-1α mRNA expression, protein stability and transactivation ability by hyperglycemia. Furthermore, the potential therapeutic strategies for complications of diabetes related to HIF-1α defects are discussed. Some of these mechanisms remain to be confirmed. Moreover, the mechanisms discussed in this review do not form a comprehensive picture explaining all questions proposed in this area. Therefore, further research involving a variety of cell types and in both cell-based systems and animal models and in tissues from patients with diabetes are required in order to identify more effective therapies for ischemic and hypoxic diseases in diabetes [19]. In fact, the key target of therapies is the overexpression of HIF-1α in high glucose environment. Therefore, theoretically, all means that are sufficient to reverse hyperglycemia-induced HIF-1α deficiencies could be used as therapies: whether these approaches function by decreasing inhibited elements (PHD, ROS, MGO) or overexpressing contributing factors (GLO1) and HIF-1α per se (gene transfer). However, in practice, all strategies require strict confirmation of pharmacology and clinical trials before representing essential significance to patients with diabetes.
Abbreviations
AGEs: advanced glycation end products; Ang II: angiotensin II; ARNT: aryl hydrocarbon receptor nuclear translocator; BBB: blood-brain barrier; bHLH: basic helix-loop-helix; CaMKII: CaM-dependent protein kinase II; CBP: CREB binding protein; CHIP: carboxyl terminus of the Hsc70-interacting protein; ChRE: carbohydrate response element; ChREBP: carbohydrate response element binding protein; CoCl2: cobalt chloride; CREB: cAMP-response element binding protein; CTAD: carboxy-terminal transactivation domain; CXCR4: C-X-C chemokine receptor type 4; DFO: desferrioxamine; DMOG: dimethyloxalylglycine; EPA: eicosapentaenoic acid; EPCs: endothelial progenitor cells; EPO: erythropoietin; FIH-1: factor-inhibiting HIF-1; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GLO1: glyoxalase I; GLUT-1/3: glucose transporter 1/3; GSIS: glucose stimulation of insulin secretion; GSH: glutathione; HIF-1: hypoxia-inducible factor 1; HO-1: haem oxygenase 1; HREs: hypoxia response elements; Hsc70: heat-shock cognate protein 70; Hsp40/70: heat shock protein 40/70; IGF: insulin-like growth factor; LDH-A: lactate dehydrogenase A; MGO: methylglyoxal; MTs: metallothioneins; NO: nitric oxide; NOS: nitric oxide synthase; NTAD: amino-terminal transactivation domain; O2-: superoxide; ODD: oxygen- dependent degradation domain; PHDs: prolyl hydroxylases; RNS: reactive nitrogen species; ROS: reactive oxygen species; RPE: retinal pigment epithelial cells; SDF-1: stromal cell-derived factor-1; SOD: superoxide dismutase; TNF-α: tumor necrosis factor-α; VEGF: vascular endothelial growth factor; VHL: Von Hippel-Lindau protein.
Acknowledgements
This work was supported by research grant from the 100-Talent Introduction Programme of Sun Yat-sen University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510-5514
2. Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. 1996;271:C1172-C1180
3. Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypox-ia-inducible transcription factor depends primarily upon re-dox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996;271:32253-32259
4. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ. et al. Targeting of HIF-1α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468-472
5. Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. 2004;279:40337-40344
6. Weidemann A, Johnson RS. Biology of HIF-1α. Cell Death Differ. 2008;15:621-627
7. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME. et al. The tumor suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271-275
8. Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1α and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1 α. Mol Cell. 2007;25:207-217
9. Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26:2019-2028
10. Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the tran-scriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466-1471
11. Semenza GL. Life with oxygen. Science. 2007;318:62-64
12. Semenza GL. Vascular responses to hypoxia and ischemia. Arterioscler Thromb Vasc Biol. 2010;30:648-652
13. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P. et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase. A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529-32537
14. Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393-402
15. Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K. et al. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: Their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999;18:1905-1914
16. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447-5454
17. Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J. et al. Genome-wide association of HIF-1α and HIF-2α DNA-binding with expression profiling of hypoxia inducible transcripts. J Biol Chem. 2009;284:16767-16775
18. Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS. et al. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A. 2009;106:4260-4265
19. Bento CF, Pereira P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia. 2011;54:1946-1956
20. Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL. et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem. 1997;272:5375-5381
21. Palmer LA, Semenza GL, Stoler MH, Johns RA. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am J Physiol. 1998;274:L212-L219
22. Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun. 1998;245:894-899
23. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858-864
24. Zagzag D, Krishnamachary B, Yee H, Okuyama H, Chiriboga L, Ali MA. et al. Stromal cell-derived factor-1 α and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res. 2005;65:6178-6188
25. Malhotra R, Tyson DG, Sone H, Aoki K, Kumagai AK, Brosius FC 3rd. Glucose uptake and adenoviral mediated GLUT1 infection decrease hypoxia-induced HIF-1alpha levels in cardiac myocytes. J Mol Cell Cardiol. 2002;34:1063-1073
26. Pillai R, Huypens P, Huang M, Schaefer S, Sheinin T, Wettig SD. et al. Aryl hydrocarbon receptor nuclear translocator/hypoxia-inducible factor-1{beta} plays a critical role in maintaining glucose-stimulated anaplerosis and insulin release from pancreatic {beta}-cells. J Biol Chem. 2011;286:1014-1024
27. Wang XL, Suzuki R, Lee K, Tran T, Gunton JE, Saha AK. et al. Ablation of ARNT/HIF1beta in liver alters gluconeogenesis, lipogenic gene expression, and serum ketones. Cell Metab. 2009;9:428-439
28. Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ. et al. Hypoxia-inducible factor-1 alpha regulates beta-cell function in mouse and human islets. J Clin Invest. 2010;120:2171-2183
29. Vordermark D, Kraft P, Katzer A, Bölling T, Willner J, Flentje M. Glucose requirement for hypoxic accumulation of hypoxia-inducible factor-1alpha (HIF-1alpha). Cancer Lett. 2005;230:122-133
30. Staab A, Löffler J, Said HM, Katzer A, Beyer M, Polat B. et al. Modulation of glucose metabolism inhibits hypoxic accumulation of hypoxia-inducible factor-1alpha (HIF-1alpha). Strahlenther Onkol. 2007;183:366-373
31. Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519-9525
32. Kalapos MP. Methylglyoxal in living organisms: chemistry, biochemistry, toxicology and biological implications. Toxicol Lett. 1999;110:145-175
33. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813-820
34. Ceradini DJ, Yao D, Grogan RH, Callaghan MJ, Edelstein D, Brownlee M. et al. Decreasing intracellular superoxide corrects defective ische-mia-induced new vessel formation in diabetic mice. J Biol Chem. 2008;283:10930-10938
35. Bento CF, Fernandes R, Matafome P, Sena C, Seica R, Pereira P. Methylglyoxal-induced imbalance in the ratio of vascular endothelial growth factor to angiopoietin 2 secreted by retinal pigment epithelial cells leads to endothelial dysfunction. Exp Physiol. 2010;95:955-970
36. Kajiwara H, Luo ZY, Belanger AJ, Urabe A, Vincent KA, Akita GY. et al. A hypoxic inducible factor-1 α hybrid enhances collateral development and reduces vascular leakage in diabetic rats. J Gene Med. 2009;11:390-400
37. Thangarajah H, Yao D, Chang EI, Shi Y, Jazayeri L, Vial IN. et al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc Natl Acad Sci U S A. 2009;106:13505-13510
38. Bento CF, Fernandes R, Ramalho J, Marques C, Shang F, Taylor A. et al. The chaperone-dependent ubiquitin ligase CHIP targets HIF-1α for degradation in the presence of methylglyoxal. PLoS ONE. 2010;5:e15062
39. Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405-412
40. Zou AP, Cowley AW Jr. Reactive oxygen species and molecular regulation of renal oxygenation. Acta Physiol Scand. 2003;179:233-241
41. Bullock JJ, Mehta SL, Lin Y, Lolla P, Li PA. Hyperglycemia-enhanced ischemic brain damage in mutant manganese SOD mice is associated with suppression of HIF-1alpha. Neurosci Lett. 2009;456:89-92
42. Muranyi M, Ding C, He Q, Lin Y, Li PA. Streptozotocin-induced diabetes causes astrocyte death after ischemia and reperfusion injury. Diabetes. 2006;55:349-355
43. Kim GW, Chan PH. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2002;22:798-809
44. Thomas DD, Ridnour LA, Espey MG, Donzelli S, Ambs S, Hussain SP. et al. Superoxide fluxes limit nitric oxide-induced signaling. J Biol Chem. 2006;281:25984-25993
45. Kimura H, Weisz A, Kurashima Y, Hashimoto K, Ogura T, D'Acquisto F. et al. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood. 2000;95:189-197
46. Hirota K, Semenza GL. Rac1 activity is required for the activation of hypoxia-inducible factor 1. J Biol Chem. 2001;276:21166-21172
47. Marfella R, D'Amico M, Di Filippo C, Piegari E, Nappo F, Esposito K. et al. Myocardial infarction in diabetic rats: role of hyperglycaemia on infarct size and early expression of hypoxia-inducible factor 1. Diabetologia. 2002;45:1172-1181
48. Botusan IR, Sunkari VG, Savu O, Catrina AI, Grünler J, Lindberg S. et al. Stabilization of HIF-1α is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci U S A. 2008;105:19426-19431
49. Mace KA, Yu DH, Paydar KZ, Boudreau N, Young DM. Sustained expression of Hif-1α in the diabetic environment promotes angiogenesis and cutaneous wound repair. Wound Repair Regen. 2007;15:636-645
50. Albina JE, Mastrofrancesco B, Vessella JA, Louis CA, Henry WL Jr, Reichner JS. HIF1 expression in healing wounds: HIF-1alpha induction in primary inflammatory cells by TNF-alpha. Am J Physiol Cell Physiol. 2001;281:C1971-7
51. Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999;59:3915-3918
52. Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1α/ARNT. EMBO J. 1998;17:5085-5094
53. Pagé EL, Robitaille GA, Pouysségur J, Richard DE. Induction of hypoxia-inducible factor-1α by transcriptional and translational mechanisms. J Biol Chem. 2002;277:48403-48409
54. Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes. 2004;53:3226-3232
55. Tamarat R, Silvestre JS, Huijberts M, Benessiano J, Ebrahimian TG, Duriez M. et al. Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc Natl Acad Sci U S A. 2003;100:8555-8560
56. Sandau KB, Fandrey J, Brüne B. Accumulation of HIF-1α under the influence of nitric oxide. Blood. 2001;97:1009-1015
57. Fukuda R, Kelly B, Semenza GL. Vascular endothelial growth factor gene expression in colon cancer cells exposed to prostaglandin E2 is mediated by hypoxia-inducible factor 1. Cancer Res. 2003;63:2330-2334
58. Xiao Q, Zeng S, Ling S, Lv M. Upregulation of HIF-1alpha and VEGF expression by elevated glucose concentration and hypoxia in cultured human retinal pigment epithelial cells. J Huazhong Univ Sci Technolog Med Sci. 2006;26:463-465
59. Yan JQ, Zhang ZY, Shi HL. HIF-1 is involved in high glucose-induced paracellular permeability of brain endothelial cells. Cell Mol Life Sci. 2012;69:115-128
60. Bensellam M, Duvillié B, Rybachuk G, Laybutt DR, Magnan C, Guiot Y. et al. Glucose-induced O2 consumption activates hypoxia inducible factors 1 and 2 in rat insulin-secreting pancreatic beta-cells. PLoS One. 2012;7:e29807
61. Puri S, Cano DA, Hebrok M. A role for von Hippel-Lindau protein in pancreatic beta-cell function. Diabetes. 2009;58:433-441
62. Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4:107-110
63. Isoe T, Makino Y, Mizumoto K, Sakagami H, Fujita Y, Honjo J. et al. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. 2010;78:48-59
64. Li J, Zhao SZ, Wang PP, Yu SP, Zheng Z, Xu X. Calcium mediates high glucose-induced HIF-1α and VEGF expression in cultured rat retinal Müller cells through CaMKII-CREB pathway. Acta Pharmacol Sin. 2012;33:1030-1036
65. Köhl R, Zhou J, Brüne B. Reactive oxygen species attenuate ni-tric-oxide-mediated hypoxia-inducible factor-1alpha stabilization. Free Radic Biol Med. 2006;40:1430-1442
66. Guo S, Bragina O, Xu Y, Cao Z, Chen H, Zhou B. et al. Glucose upregulates HIF-1 alpha expression in primary cortical neurons in response to hypoxia through maintaining cellular redox status. J Neurochem. 2008;105:1849-1860
67. Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T. et al. Hyperglycaemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801-813
68. Chowdhury R, Hardy A, Schofield CJ. The human oxygen sensing machinery and its manipulation. Chem Soc Rev. 2008;37:1308-1319
69. Goldberg MA, Dunning SP, Bunn HF. Regulation of the erythropoietin gene: Evidence that the oxygen sensor is a heme protein. Science. 1988;242:1412-1415
70. Labbe RF, Hubbard N. Metal specificity of the iron-protoporphyrin chelating enzyme from rat liver. Biochim Biophys Acta. 1961;52:130-135
71. Ohtomo S, Nangaku M, Izuhara Y, Takizawa S, Strihou CY, Miyata T. Cobalt ameliorates renal injury in an obese, hypertensive type 2 diabetes rat model. Nephrol Dial Transplant. 2008;23:1166-1172
72. Hirota K, Semenza GL. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem Biophys Res Commun. 2005;338:610-616
73. Trivin F, Beaumont C, Emerit J. Iron metabolism, free radicals, and oxidative injury. Biomed Pharmacother. 2001;55:333-339
74. Yang P, Reece EA. Role of HIF-1α in maternal hyperglycemia-induced embryonic vasculopathy. Am J Obstet Gynecol. 2011;204:332.e1-7
75. Taneda S, Honda K, Tomidokoro K, Uto K, Nitta K, Oda H. Eicosapentaenoic acid restores diabetic tubular injury through regu-lating oxidative stress and mitochondrial apoptosis. Am J Physiol Renal Physiol. 2010;299:F1451-F1461
76. Cai L, Wang Y, Zhou G, Chen T, Song Y, Li X. et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;48:1688-1697
77. Feng W, Wang Y, Cai L, Kang YJ. Metallothionein rescues hypox-ia-inducible factor-1 transcriptional activity in cardiomyocytes under diabetic conditions. Biochem Biophys Res Commun. 2007;360:286-289
78. Thornalley PJ. Glyoxalase I - structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans. 2003;31:1343-1348
79. Dinçer Y, Alademir Z, Ilkova H, Akçay T. Susceptibility of glutatione and glutathione-related antioxidant activity to hydrogen peroxide in patients with type 2 diabetes: effect of glycemic control. Clin Biochem. 2002;35:297-301
Author contact
![]() Corresponding author: Tongfeng Zhao, PhD. Department of Endocrinology, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou 510655, China. E-mail: zhaotongfengcom.
Corresponding author: Tongfeng Zhao, PhD. Department of Endocrinology, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou 510655, China. E-mail: zhaotongfengcom.