Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Material and Methods
3. Results
4. Discussion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(10):1278-1285. doi:10.7150/ijms.6254 This issue Cite
Research Paper
Sugammadex, a Neuromuscular Blockade Reversal Agent, Causes Neuronal Apoptosis in Primary Cultures
José M. Palanca1, Diana Aguirre-Rueda2, Manuel V. Granell1, Martin Aldasoro2, Alma Garcia2, Antonio Iradi2, Elena Obrador2, Maria Dolores Mauricio2, Jose Vila2, Anna Gil-Bisquert2, Soraya L. Valles2 ![]()
1. Department of Surgery/Anaesthesiology, School of Medicine and Odontology, University of Valencia, Spain;
2. Department of Physiology, School of Medicine and Odontology, University of Valencia, Spain.
Received 2013-3-15; Accepted 2013-7-18; Published 2013-8-3
Abstract
Sugammadex, a γ-cyclodextrin that encapsulates selectively steroidal neuromuscular blocking agents, such as rocuronium or vecuronium, has changed the face of clinical neuromuscular pharmacology. Sugammadex allows a rapid reversal of muscle paralysis. Sugammadex appears to be safe and well tolerated. Its blood-brain barrier penetration is poor (< 3% in rats), and thus no relevant central nervous toxicity is expected. However the blood brain barrier permeability can be altered under different conditions (i.e. neurodegenerative diseases, trauma, ischemia, infections, or immature nervous system).
Using MTT, confocal microscopy, caspase-3 activity, cholesterol quantification and Western-blot we determine toxicity of Sugammadex in neurons in primary culture. Here we show that clinically relevant sugammadex concentrations cause apoptotic/necrosis neuron death in primary cultures. Studies on the underlying mechanism revealed that sugammadex-induced activation of mitochondria-dependent apoptosis associates with depletion of neuronal cholesterol levels. Furthermore SUG increase CytC, AIF, Smac/Diablo and CASP-3 protein expression in cells in culture. Potential association of SUG-induced alteration in cholesterol homeostasis with oxidative stress and apoptosis activation occurs. Furthermore, resistance/sensitivity to oxidative stress differs between neuronal cell types.
Keywords: Sugammadex, apoptosis, CytC, AIF, Smac/Diablo and CASP-3.
1. Introduction
Sugammadex (SUG), a γ-cyclodextrin with a doughnut-like molecular structure containing a lipophilic core and a hydrophilic periphery, is the first in a new (and revolutionary) class of selective binding reversal agents that terminates neuromuscular blockade (NMB) with different NMB drugs such as rocuronium (ROC) or vecuronium (VEC) [1]. The negative charges of the external chains maintain the cavity open and the core, with Van Der Waals forces, thermodynamic bonds and hydrophobic interactions, creates a 1:1 ratio complex with very tight bonds between the NMB drug and SUG [2]. SUG, depending on the dose, can reverse a moderate or deep NMB without muscle weakness [3].
A Cochrane systematic review including 18 randomized controlled trials (with a total of 1321 patients) on the efficacy and safety of SUG concluded that it was more effective than placebo (no medication) or neostigmine in reversing muscle relaxation caused by NMB during surgery and is relatively safe. Serious complications occurred in less than 1% of the patients who received SUG [4]. Reported side effects for SUG included cough, dry mouth, temperature changes, parasomia, paresthesia, movement during surgery, mild erythemia, abdominal discomfort, tachycardia, bradycardia, dizziness, increased creatinine phosphokinase, and increased B2 microglobinuria [3]. Pooled preclinical safety data obtained from the US FDA briefing document [5]; and the European Medicines Agency scientific document (EMEA)[6] reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and toxicity to reproduction, local tolerance or compatibility with blood. Potential kidney and lung toxicity of cyclodextrins, if large repeated doses are administered, must be also taken into account [7]. Besides, available information regarding SUG on special conditions/population, including renal impairment, elderly patients, obese patients, and pediatric population is really limited [5, 6, 8].
Owing to its singular architecture, charge, and high molecular weight (2178.01), SUG exhibits a very low transfer across the blood-brain barrier (BBB) and the placenta [9]. This particularity apparently precludes any significant SUG-derived toxicity in the central nervous system (CNS). Nevertheless, different clinical conditions imply moderate or severe alterations of the BBB integrity, i.e. Alzheimer [10], Parkinson [11] or multiple sclerosis [12] disease-associated neurodegeneration, traumatic brain/spinal cord injury [13], ischemia [14], meningitis [15], or immature nervous system [16]. Under these clinical conditions, SUG may cross the BBB in specific areas. This report suggests, for the first time, potential SUG-induced neuronal toxicity.
2. Material and Methods
2.1. Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) was obtained from Gibco life technology (Gibco Invitrogen Corporation, Barcelona, Spain). SUG was from Merck Millipore products, Sharp & Dohme (Madrid, Spain). Appropriate concentration of SUG was tested in figure 1 and selected a standard concentration of 75 µg/ml during 24 h for further experiments. Western blot enhanced chemo-luminescent detection system (ECL) was from Amersham Bioscience (Amersham Biosciences, Barcelona, Spain). Monoclonal anti-glial fibrillary acidic protein (anti-GFAP) antibody (1:500) was from Sigma-Aldrich Biotech (Madrid, Spain). Monoclonal anti-MAP-2 protein (anti-MAP-2) antibody (1:500) and monoclonal anti-cytochrome C protein (anti-CytC) antibody (1:500) were from Santa Cruz Biotechnology (Barcelona, Spain). Anti-apoptosis-inducing factor (anti-AIF) (1:500) and anti-Smac/Diablo (1:500) were from Sigma-Aldrich Biotech. (Barcelona, Spain). All other reagents were of analytical or culture grade purity.
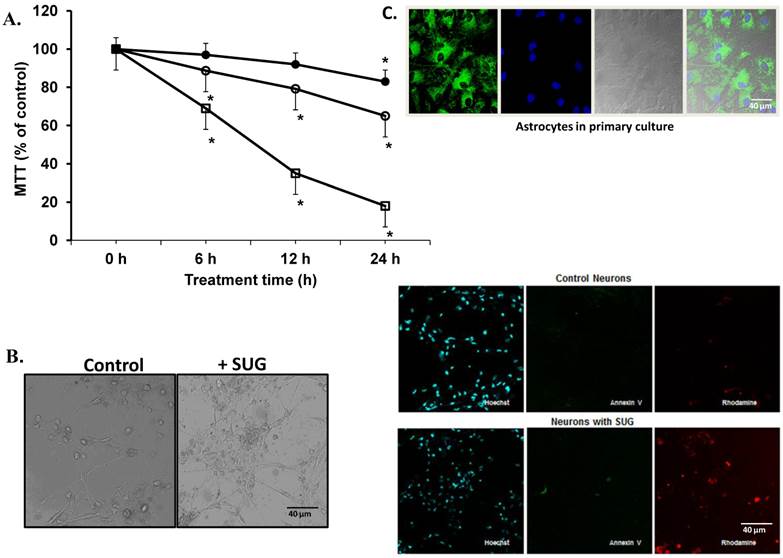
(A) Neuronal cell death induced by sugammadex. Time- and concentration-dependent neurotoxicity of SUG was determined by the MTT assay (*p < 0.01). ● (37.5 µg SUG/ml), ○ (75 µg SUG/ml), and □ (150 µg SUG/ml). Data are means ± SD for 5 independent experiments. *p < 0.01 comparing SUG vs controls (no additions). ANOVA (two way) was used when needed. (B) Optic microscopic image of cultured neurons incubated in the absence or in the presence of SUG (75 µg/ml, 24 h). The % of viable cells in control cultures was always > 95%. (C) Immunofluorescence of neurons in primary culture with Hoechst, Annexin V and Rhodamine was used. Astrocytes marked with Hoechst and mitotracker also are shown.
2.2. Primary cultures
Primary cultures of cortical neurons were prepared from the cerebral cortex (5 female rats to obtain the fetuses) of 14-15-day old rat fetuses. Briefly the cerebral cortex, obtained under toxin-free sterile conditions, was dissected and dissociated mechanically by pipetting 10 times with DMEM (10 ml for the cortex obtained from 12-14 fetuses). The cell suspension was filtered through a nylon mesh with a pore size of 90 μm. Cell suspension was plated (5 x 104 cells/cm2) on poly-lysine-coated dishes. After attachment of the cells (1 h), the plating medium was changed to DMEM pH 7.4, supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 40 mM NaHCO3, 100 units/ml penicillin, and 100 mg/ml streptomycin. Primary cultures of cortical astrocytes were prepared from the cerebral cortex (5 female rats to obtain the fetuses) of 21-day old rat fetuses. Briefly the cerebral cortex, obtained under toxin-free sterile conditions, was dissected and dissociated mechanically by pipetting 10 times with DMEM (10 ml for the cortex obtained from 12-14 fetuses). The cell suspension was filtered through a nylon mesh with a pore size of 90 μm. Cell suspension was plated (5 x 104 cells/cm2) with 20% FBS, 10 mM HEPES, 40 mM NaHCO3, 100 units/ml penicillin, and 100 mg/ml streptomycin. After 4 days, the plating medium was changed with medium prepared such as above. At seven days of culture 20% was changed to 10% FBS.
Cultures were grown in a humidified atmosphere of 5% CO2 and 95% O2, at 37oC, for 3 days, and then exposed to cytosine β-D arabinofuranoside (10 μM) for 24 h to prevent proliferation of no neuronal cells. Possible contamination by astrocytes was assessed by immune-fluorescence using monoclonal anti-glial fibrillary acidic protein (GFAP) (astrocytes marker: Sigma-Aldrich. 1:500) antibody (clone G-A-5) and polyclonal anti-microtubule-associated protein 2 (MAP-2) (neuronal marker: Sigma-Aldrich. 1:500). Under these conditions approx. 99% ± 3 of all cultured cells were neurons [17]. Cell viability, assessed by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay [18], was normalized relative to control cells whose viability was taken as 100% when treated for the indicated periods with the corresponding vehicles. Cell viability in controls, as measured by the release of lactate dehydrogenase activity to the extracellular medium, was always >90%. All animal work was performed according to minimize animals suffering and to reduce the number of animals used following the local ethics committees (Ethics Committee for Animal Research of the University of Valencia) based on the local guidelines (Spanish law 32/2007).
2.3. Immunofluorescence and confocal microscopy
Neurons monolayers growing on 16-mm glass coverslips were used for immune-fluorescence studies following the procedure described by Valles et al. [17]. Monoclonal anti-MAP-2 and anti-GFAP antibodies (Sigma-Aldrich) were used to see the purity of culture. Incubation was performed at 37ºC for 60 min. After washing several times with phosphate-buffered saline (PBS), neurons were then incubated with secondary antibodies. When necessary, the nuclei were stained (0.5 µg/ml) with Hoechst 33342 dye (Molecular Probes) for 5 min. In other experiments, astrocytes were incubated with mitotracker and Hoechst and neurons were incubated simultaneously with Annexin V, Hoechst and Rhodamine to see apoptosis and necrosis.
2.4. Cell death analysis
Apoptotic and necrotic cell death induced by SUG were distinguished by using fluorescence microscopy [19]. Briefly, isolated cells were incubated with Hoescht 33342 (10 mM; which stains all nuclei) and Rhodamine (10 µg/ml; to stain mitochondria in living cells), for 3 min, and analyzed using a Diaphot 300 fluorescence microscope (Nikon, Tokyo, Japan) with excitation at 360 nm. Nuclei of viable, necrotic, for apoptotic cells were observed as blue round nuclei, pink round nuclei, and fragmented blue or pink nuclei, respectively. About 1500 cells were counted each time. DNA strand breaks in apoptotic cells were assayed by using a direct TUNEL labeling assay (Boehringer, Mannheim, Germany) and fluorescence microscopy following manufacturer's methodology.
2.5. Western-blots
Attached cells (neurons in primary culture) was collected and were harvested with ice-cold buffer (0.125 M Tris-HCl, pH 6.8, 2% SDS, 19% glycerol, 1 mM phenylmethylsulfonyl fluoride, 1μg/ml leupeptin, 1μg/ml aprotinin, and 1μg/ml pepstatin), pH 7.4, disrupted by repeated cycles of freezing and thawing, and boiled for 5 min at 90º. Cell extracts were homogenized and centrifuged (10000 x g for 10 min at 4ºC). The resulting supernatant was used for western-blot analysis. Protein concentration was determined using a modified Lowry method [20]. Proteins were separated by SDS-PAGE gels and transferred to nitrocellulose membranes using standard techniques. Membranes were blocked with 5% dried milk in TBS containing 0.05% Tween-20 and then incubated with the corresponding antibodies following manufacturer's recommendations. The blots were washed three times with phosphate-buffered saline containing 0.2% Tween 20 for 15 min and then incubated for 1 h with a secondary horseradish peroxidase-linked anti-rabbit or anti-mouse IgG antibody (Cell Signaling Technologies, Barcelona, Spain). ECL (Pharmacia biotechnology, San Francisco, CA) was used for developing. Auto-radiographic signals were assessed using a Bio-Rad scanning densitometer.
2.6. TUNEL assay
Apoptotic cells were assessed by measuring DNA fragmentation in a standard TUNEL assay following the instructions with the kit. In brief, cells were permeabilized with 0.1% Triton X-100 for 2 min on ice, reacted with TdT enzyme, and stained with FITC-dUTP for 1 h at 37°C in dark. The TUNEL-positive cells were imaged under a fluorescent microscope and the cells with green fluorescence were defined as apoptotic cells.
2.7. Caspase 3 activity assay
This activity was measured in cytosolic fractions (see below) by using a highly sensitive colorimetric substrate, N-acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) following manufacturer's instructions (CalBiochem, La Jolla, CA). Enzyme activity was calculated, using manufacturer's formulae, as pmol/min.
2.8. Sub-cellular fractionation and cholesterol quantification
Cultured cortical neurons underwent fractionation to segregate cytosol and membrane fractions as previously described [21, 22]. Briefly, cells in the supernatant collect by centrifugation and attached cells scraped in 5 mm EDTA in PBS were pelleted by centrifugation for 10 min at 1000 x g at 4°C. Pellets were re-suspended in 200 μl of fractionation buffer (0.25 m sucrose, 1 mM Mg chloride, 2 mM EGTA, and 25 mM HEPES, pH 7.4) and lysed by three cycles of flash freezing in liquid nitrogen. Lysates were then centrifuged at 100000 × g for 30 min at 4oC. Supernatants (cytosol fraction) were separated and the pellet (membrane fraction) was dissolved again in 200 μl of fractionation buffer containing 5% Triton X-100 and briefly sonicated. Whole-cell lysates were obtained by harvesting or homogenizing the samples in sub-cellular fractionation buffer containing 2.5% Triton X-100, followed by brief sonication.
Neurons were cultured, as indicated above, with or without increasing amounts of soluble cholesterol (see Results) and/or SUG. Whole cell lysates and sub-cellular fractions were lyophilized using freeze-drying equipment from SP Scientific (Gardiner, NY). Lipids were extracted and cholesterol was determined using cholesterol oxidase as previously described [23].
2.9. Statistical significance
Statistical analyses were performed using t-test (Student's t test) and also a one way ANOVA was performed with the program GraphPad Prism, with a p < 0.05 being considered significant.
3. Results
3.1. Sugammadex causes neuronal cell death
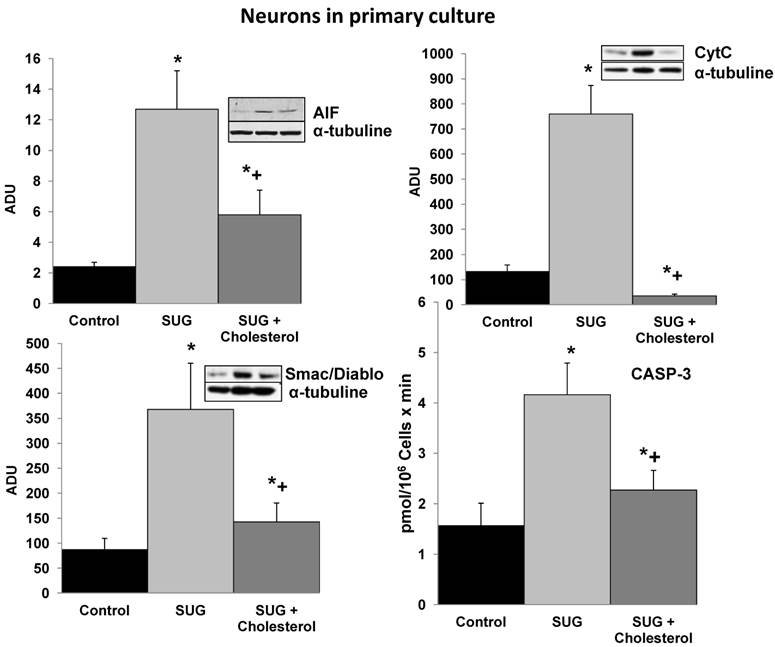
Based on the results displayed in Figure 1, we selected a standard concentration of 75 µg/ml of SUG for further experiments. Also using immune-fluorescence technique with Anexine V (apoptosis) and Rhodamine (necrosis), we detected an increase in apoptosis and necrosis after SUG addition. In the experiments displayed as Table 1, we found no significant differences when the sum of dead cells (calculated along the culture time) and viable cells obtained in the presence or in the absence of sugammadex was compared (not shown). However, the total number of dead neurons was much higher in the presence of the γ-cyclodextrin (Table 1), which means that neuronal death is the predominant effect caused by sugammadex. Most dying cells (90%) displayed apoptotic features and only a small percentage was identified as necrotic (Table 1). The percentage of apoptotic cells obtained by using Hoechst 33342 and propidium iodide (Table 1) or the TUNEL technique (not shown) was similar. During the 48 h period in the presence of the γ-cyclodextrin, almost all nonviable cells (94.7 + 3.9 %) lost their attachment to the flasks and were progressively recovered in the culture medium. Looking for different pro-apoptotic molecular signals (using Western-blot technique), we demonstrated in figure 2 increase protein expression in CytC, AIF and Smac/Diablo after SUG addition compared with control cells. Also higher activity of CASP-3 activity was noted comparing SUG with control samples (Figure 2).
Type of neuronal cell death induced by sugammadex.
| Parameter | No addition | SUG |
|---|---|---|
| 10-4 x number of death cells/ml | 2.1 ± 0.7 | 8.3 ± 2.3* |
| Necrotic cells (%) | 46.6 ± 7.5 | 12.3 ± 4.4* |
| Apoptotic cells (%) | 53.4 ± 9.3 | 87.7 ± 10.6* |
Cholesterol prevents the increase in pro-apoptotic molecular signals induced by Sugammadex. SUG (75 µg/ml) was present in the culture medium for 24 h. CytC, AIF, and Smac/Diablo (western blots) and CASP-3 activity were analyzed as indicated under Materials and Methods. Results of quantitative analysis of immunoblots are means ± SD (error bars) (n=5) expressed as relative changes in arbitrary densitometry units (ADU) normalized against an internal standard (α-tubulin). *p < 0.01 comparing all conditions vs controls (no additions); +p < 0.01 comparing cholesterol + SUG vs SUG.
3.2. Sugammadex-induced activation of apoptosis is linked to an alteration of cellular cholesterol homeostasis
To further investigate the underlying mechanisms responsible of the SUG-induced apoptosis activation, we evaluated if this γ-cyclodextrin could cause alterations in neuronal cholesterol levels and whether this could be linked to apoptosis. As shown in Table 2, sub-cellular fractionation revealed that SUG indeed decreased membrane-associated, cytosolic, and mitochondrial cholesterol levels. Moreover, as shown in Figure 2, cholesterol enrichment prevented the release of different pro-apoptotic molecular signals (including CytC, AIF, Smac/Diablo, or CASP-3 activity) caused by SUG. Thus, indicating a relationship between alteration of neuronal cholesterol homeostasis and apoptosis activation induced by SUG addition to neuronal cells.
Effect of sugammadex on the cellular content of Cholesterol in neurons, astrocytes, endothelial cells, and leukocytes.
| Cholesterol (µg/mg protein) | MTT (% control) | |||||
|---|---|---|---|---|---|---|
| Whole cell | Plasma membrane | |||||
| - SUG | +SUG | -SUG | +SUG | -SUG | +SUG | |
| Neurons | 30.3 ± 3.7 | 18.5 ± 2.7* | 67.4 ± 5.9 | 35.1 ± 4.7* | 100 ± 2 | 65 ± 7* |
| Astrocytes | 51.7 ± 8.2 | 40.1 ± 5.3 | 215.6 ± 26.5 | 177.4 ± 10.2* | 100 ± 8 | 92 ± 10 |
| Endoth. cells | 42.4 ± 3.9 | 31.7 ± 3.9* | 137.3 ± 19.2 | 116.7 ± 8.7* | 100 ± 3 | 99 ± 12 |
| Leukocytes | 29.6 ± 4.4 | 20.1 ± 3.5* | 176.6 ± 15.2 | 139.3 ± 9.5* | 100 ± 9 | 97 ± 13 |
Effect of plasma cholesterol-enrichment on sugammadex-induced neuron cell death.
| Treatment | Plasma Membrane Cholesterol (µg/mg protein) | MTT (% Control) |
|---|---|---|
| None | 62.2 ± 3 | 100 ± 3 |
| SUG | 36.6 ± 5.1* | 63 ± 6* |
| Cholesterol | 83.7 ± 7.9* | 102 ± 2 |
| SUG + Cholesterol | 60.5 ± 6.9 | 97 ± 3 |
4. Discussion
NMB drugs are currently used during surgery to facilitate muscle relaxation for endotracheal intubation and artificial ventilation, ensure patient immobility, and easier surgical access to body cavities [24].
The main advantage of SUG is reversal of NMB without relying on inhibition of acetylcholinesterase. The amino-steroid molecule bound within SUG's lipophilic core is rendered unavailable to bind to the acetylcholine receptor at the neuromuscular junction [25]. This avoids the autonomic instability produced by anti-cholinesterase drugs such as neostigmine, and thus antimuscarinic agents such as atropine do not need to be co-administered. Moreover, when a fast onset and short duration of muscle relaxant is required, there has been little choice previously apart from the use of suxamethonium. Nevertheless, suxamethonium is not ideal since it has some undesirable side effects, such as anaphylaxis, increasing serum potassium levels and different cardiovascular responses. Since, in high doses, ROC has a reasonably rapid onset and now can be reversed with SUG, it can potentially be used instead [26].
The present results indicate potential neuronal toxicity at clinically relevant doses of SUG. Nevertheless, as stated in the Introduction, these harmful effects should be limited to conditions associated with moderate or severe alterations of the BBB integrity. It is in this specific context where our findings may represent a warning, particularly when relatively high doses of this γ-cyclodextrin are required for the rapid reversal of NMB. SUG, which does not bind to plasma proteins or erythrocytes, distributes into the extracellular water of the body, being its distribution volume in adults of approximately 11-14 l [6, 26]. Single-dose administration of SUG 16, 20, or 32 mg/kg in combination with ROC 1.2 mg/kg or VEC 0.1 mg/kg was well tolerated in healthy volunteers with no clinical evidence of residual NMB, thus concluding that these combinations can safely be administered simultaneously [27]. Nevertheless, because SUG forms a 1:1 complex with steroidal NMB drugs, most of the SUG molecules administered remain free in the extracellular water. As shown in Figure 1.A, SUG decreases neuronal cell viability in a concentration- and time-dependent fashion. Pharmacokinetic studies in healthy volunteers showed that after simultaneous i.v. administration of SUG (16 mg/kg) and ROC (1.2 mg/kg) or VEC (0.1 mg/kg), the peak plasma concentration of the unbound γ-cyclodextrin was of 70-80 mg/ml [27].
SUG causes cell death, predominantly by apoptosis, in cultured neurons (Figure 1, Table 1). Apoptosis induction associates with an alteration in neuronal cholesterol homeostasis (Table 2). Interestingly, it has been shown in A431 cancer cells that cholesterol depletion using methyl-β-cyclodextrin causes apoptosis [28]; and in human keratinocytes that disruption of lipid rafts by cholesterol-depleting compounds (methyl-β-cyclodextrin, filipin III, cholesterol oxidase, or mevastatin) leads to a spontaneous clustering of Fas in the non-raft compartment of the plasma membrane, formation of Fas-FADD complexes, activation of caspase-8, and apoptosis [29]. In fact, neuronal death caused by inhibition of intracellular cholesterol trafficking has been shown to be caspase dependent and associated with activation of the mitochondrial apoptosis pathway [30]. Cholesterol is an abundant component of plasma membranes of eukaryotic cells and is an essential regulator of membrane fluidity, permeability, receptor function, and ion channel activity [31]. In fact maintenance of its cellular homeostasis is needed to support cell proliferation and survival [32]. The distribution of cholesterol in the membranes is not uniform and its content is particularly high in the submicroscopic areas also enriched in gangliosides and sphingolipids. These micro-domains known as lipid rafts, act as molecular platforms that spatially organize membrane receptor molecules [33]. β-cyclodextrin and carboxymethyl-β-cyclodextrin induced caspase-dependent apoptotic cell death in human keratinocytes on depletion of membrane cholesterol; whereas α-cyclodextrin, γ-cyclodextrin, and 2-hydroxypropyl-β-cyclodextrin were not apoptotic to this type of cell [34]. Moreover, it is reported that cholesterol depletion by methyl-β-cyclodextrine blocks cholera toxin transport from endosomes to the Golgi apparatus in hippocampal neurons [35]; and that enrichment of the neuronal plasma membrane with cholesterol protects cortical neurons from apoptosis induced by soluble oligomers of the Aβ(1-40) peptide [36]. The present report shows for the first time that a γ-cyclodextrin can cause neuronal damage, and that this effect associates with cholesterol depletion, and that it can be prevented by preloading the neurons with cholesterol (Figure 2).
Some reports support indeed the idea that cholesterol may actually be good for the brain. Consistent with this, for instance, maintenance of physiological levels of cellular cholesterol appears to decrease the neuronal death (reactive oxygen species-associated) caused by prion peptides through regulation of p53, P-p38 and P-ERK protein levels [37]. Oxidative stress plays an important role in neurodegenerative diseases, and recent results show that elevated cellular cholesterol may prevent oxidative stress in different neurodegenerative diseases including Alzheimer's disease [38]. Moreover, adaptation of neuronal cells to chronic oxidative stress is associated with cholesterol accumulation and altered sphingolipid homeostasis and lysosomal function [39].
Cell stress-associated mitochondrial CytC release is largely caspase independent, where release of Smac/Diablo is a caspase-catalysed event that occurs downstream of CytC release. Furthermore, the literature suggests that apoptosis-associated CytC and Smac/Diablo release from mitochondria do not occur via the same mechanism. Smac/Diablo export from mitochondria into the cytosol is provoked by cytotoxic drugs and DNA damage, as well as by ligation of the CD95 death receptor [30]. Figure 2 demonstrate a higher Smac/Diablo protein expression after SUG addiction compared with control samples, showing a cell death caused by inhibition of intracellular cholesterol trafficking caspase dependent and associated with activation of the mitochondrial apoptosis pathway. Furthermore, reduction of Smac/Diablo and CytC after SUG + Cholesterol was detected compared with SUG sample. Other authors have demonstrated an increase in Smac/Diablo apoptosis pathway, probably by inhibition of bone marrow B-cell development and thymic T cell differentiation and decreasing splenic T cells, such as has been published by Huang and collaborators [30].
Nevertheless cholesterol accumulation may be a double edge sword since, as other reports suggest, excessive accumulation of cholesterol in mitochondria may be a key step in promoting e.g. Alzheimer disease progression [40]. In this scenario U18666A, a cholesterol transport-inhibiting agent, leads to high intracellular cholesterol accumulation in primary cortical neurons, activation of caspases and calpains, hyperphosphorylation of tau, and apoptosis [41].
The potential association of SUG-induced alteration in cholesterol homeostasis with oxidative stress and apoptosis activation, the fact that resistance/sensitivity to oxidative stress may likely differ between brain regions and neuronal cell types, potential neurons-astrocytes interactions, as well as modulation by pathological mechanisms such as inflammation, all represent new research windows that deserve further studies.
Abbreviations
SUG, Sugammadex; CytC, Cytochrome C; ROC, Rocuronium; VEC, Vecuronium; NMB, neuromuscular blockade; AIF, apoptosis-inducing factor; and CASP-3, Caspase 3
Acknowledgements
This work was supported by a grant from the Generalitat Valenciana (GVcs2007-AP-001) (Spain) and by the University of Valencia.
Conflict of interest
Non conflict of interest is shown.
References
1. Naguib M. Sugammadex: another milestone in clinical neuromuscular pharmacology. Anesth. Analg. 2007;104:575-581
2. Bom A Bradley M, Cameron K. et al. A novel concept of reversing neuromuscular block: Chemical. encapsulation of rocuronium bromide by a cyclodextrin-based synthetic host. Angew Chem. Int. Ed Engl. 2002;41:266-270
3. Kovac AL. Sugammadex: the first selective binding reversal agent for neuromuscular block. J. Clin Anesth. 2009;21:444-453
4. Abrishami A, Ho J, Wong J. et al. Sugammadex, a selective reversal.medication for preventing postoperative residual neuromuscular blockade. Cochrane Database. Syst Rev. 2009:CD007362
5. Anesthetic and Life Support Advisory Committee Meeting: briefing document (background package) for sugammadex sodium injection [online]. Food and Drug Administration. http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008-4346b1-02-Organon.pdf
6. European Public Assessment Report for Bridion (sugammadex): scientific discussion [online]. European Medicines Agency. http://www.emea.europa.eu/humandocs/PDFs/EPAR/bridion/H-885-en6.pdf
7. Laza-Knoerr AL, Gref R, Couvreur P. Cyclodextrins for drug delivery. Drug Target. 2010;18:645-656
8. Akha AS, Rosa J, Jahr JS. et al. Sugammadex: cyclodextrins, development of selective binding agents, pharmacology, clinical development, and future directions. Anesthesiol Clin. 2010;28:691-708
9. Farrar JT. Meeting of the Anesthetic and Life Support Drugs FDA Advisory Committee, Silver Spring, MD. Sugammadex. 2008:222-225
10. Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009;118:103-113
11. Stolp HB, Dziegielewska KM. Review: Role of developmental inflammation and blood-brain barrier dysfunction in neurodevelopmental and neurodegenerative diseases. Neuropathol Appl Neurobiol. 2009;35:132-146
12. Waubant E. Biomarkers indicative of blood-brain barrier disruption in multiple sclerosis. Disease Markers. 2006;22:235-44
13. Shlosberg D, Benifla M, Kaufer D. et al. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurology. 2010;6:393-403
14. Kaur C, Ling EA. Blood brain barrier in hypoxic-ischemic conditions. Curr Neurovasc Res. 2008;5:71-81
15. Mook-Kanamori BB, Geldhoff M, van der Poll T. et al. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev. 2011;24:557-591
16. Saunders NR, Knott GW, Dziegielewska KM. Barriers in the immature brain. Cell Mol Neurobiol. 2000;20:29-40
17. Valles SL, Borras C, Gambini J. et al. Oestradiol or genistein rescues neurons from amyloid beta-induced cell death by inhibiting activation of p38. Aging Cell. 2008;7:112-118
18. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55-63
19. Shimizu S, Eguchi Y, Kamiiki W. et al. Induction of apoptosis as well as necrosis by hypoxia and predominant prevention of apoptosis by Bcl-2 and Bcl-XL. Cancer Res. 1996;56:2161-2166
20. Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346-356
21. Van der Bliek AM, Redelmeier TE, Damke H. et al. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553-563
22. Kelly BL, Ferreira A. Beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281:28079-28089
23. Heider JG, Boyett RL. The picomole determination of free and total cholesterol in cells in culture. J Lipid Res. 1978;19:514-518
24. Claudius C, Garvey LH, Viby-Mogensen J. The undesirable effects of neuromuscular blocking drugs. Anaesthesia. 2009;64:10-21
25. Adam JM, Bennett DJ, Bom A. et al. Cyclodextrin-derived host molecules as reversal agents for the neuromuscular blocker rocuronium bromide: synthesis and structure-activity relationships. J Med Chem. 2002;45:1806-16
26. Yang LP, Keam SJ. Sugammadex: a review of its use in anaesthetic practice. Drugs. 2009;69:919-942
27. Cammu G, De Kam PJ, Demeyer I. et al. Safety and tolerability of single intravenous doses of sugammadex administered simultaneously with rocuronium or vecuronium in healthy volunteers. Br J Anaesth. 2008;100:373-379
28. Li YC, Park MJ, Ye SK. et al. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168:1107-1118
29. Gniadecki R. Depletion of membrane cholesterol causes ligand-independent activation of Fas and apoptosis. Biochem Biophys Res Commun. 2004;320:165-169
30. Huang Z, Hou Q, Cheung NS. et al. Neuronal cell death caused by inhibition of intracellular cholesterol trafficking is caspase dependent and associated with activation of the mitochondrial apoptosis pathway. J Neurochem. 2006;97:280-291
31. Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111-136
32. Martin M, Dotti CG, Ledesma MD. Brain cholesterol in normal and pathological aging. Biochim Biophys Acta. 2010;1801:934-944
33. Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31-39
34. Schonfelder U, Radestock A, Elsner P. et al. Cyclodextrin-induced apoptosis in human keratinocytes is caspase-8 dependent and accompanied by mitochondrial cytochrome c release. Exp Dermatol. 2006;15:883-890
35. Shogomori H, Futerman AH. Cholesterol depletion by methyl-β-cyclodextrine blocks cholera toxin transport from endosomes to the golgi apparatus in hippocampal neurons. J Neurochem. 2001;78:991-999
36. Sponne I, Fifre A, Koziel V. et al. Membrane cholesterol interferes with neuronal apoptosis induced by soluble oligomers but not fibrils of amyloidbeta peptide. FASEB J. 2004;18:836-838
37. Jeong JK, Seol JW, Moon MH. et al. Cellular cholesterol enrichment prevents prion peptide-induced neuron cell damages. Biochem Biophys Res Commun. 2010;401:516-520
38. Pensalfini A, Zampagni M, Liguri G. et al. Membrane cholesterol enrichment prevents Aβ-induced oxidative stress in Alzheimer's fibroblasts. Neurobiol Aging. 2011;32:210-222
39. Clement AB, Gamerdinger M, Tamboli IY. et al. Adaptation of neuronal cells to chronic oxidative stress is associated with altered cholesterol and sphingolipid homeostasis and lysosomal function. J Neurochem. 2009;111:669-682
40. Garcia-Ruiz C, Mari M, Colell A. et al. Mitochondrial cholesterol in health and disease. Histol Histopathol. 2009;24:117-132
41. Koh CH, Qi RZ, Qu D. et al. U18666A-mediated apoptosis in cultured murine cortical neurons: role of caspases, calpains and kinases. Cell Signal. 2006;18:1572-1583
Author contact
![]() Corresponding author: Soraya L. Valles, Ph.D. Department of Physiology, School of Medicine and Odontology, University of Valencia., 15 Av. Blasco Ibanez. 46010 Valencia, Spain. Tel: +34 963 983 813. Fax: +34 963 864 642 E-mail: lilian.valleses. Highlights: SUG causes neuronal apoptosis/necrosis.
Corresponding author: Soraya L. Valles, Ph.D. Department of Physiology, School of Medicine and Odontology, University of Valencia., 15 Av. Blasco Ibanez. 46010 Valencia, Spain. Tel: +34 963 983 813. Fax: +34 963 864 642 E-mail: lilian.valleses. Highlights: SUG causes neuronal apoptosis/necrosis.