Impact Factor ISSN: 1449-1907
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Acknowledgements
References
Author Biology

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(9):1242-1249. doi:10.7150/ijms.6541 This issue Cite
Research Paper
Arrhythmogenesis Toxicity of Aconitine Is Related to Intracellular Ca2+ Signals
Yu-hong Zhou*, Xian-mei Piao*, Xue Liu, Hai-hai Liang, Lei-min Wang, Xue-hui Xiong, Lu Wang, Yan-jie Lu, Hong-li Shan ![]()
Department of Pharmacology (the State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China
* These authors contributed equally to this study.
Received 2013-4-23; Accepted 2013-7-22; Published 2013-8-1
Abstract
Aconitine is a well-known arrhythmogenic toxin and induces triggered activities through cardiac voltage-gated Na+ channels. However, the effects of aconitine on intracellular Ca2+ signals were previously unknown. We investigated the effects of aconitine on intracellular Ca2+ signals in rat ventricular myocytes and explored the possible mechanism of arrhythmogenic toxicity induced by aconitine. Ca2+ signals were evaluated by measuring L-type Ca2+ currents, caffeine-induced Ca2+ release and the expression of NCX and SERCA2a. Action potential and triggered activities were recorded by whole-cell patch-clamp techniques. In rat ventricular myocytes, the action potential duration was significantly prolonged by 1 µM aconitine. At higher concentrations (5 µM and 10 µM), aconitine induced triggered activities and delayed after-depolarizations (6 of 8 cases), which were inhibited by verapamil. Aconitine (1 µM) significantly increased the ICa-L density from 12.77 ± 3.12 pA/pF to 18.98 ± 3.89 pA/pF (n=10, p<0.01). The activation curve was shifted towards more negative potential, while the inactivation curve was shifted towards more positive potential by 1 μM aconitine. The level of Ca2+ release induced by 10 mM caffeine was markedly increased. Aconitine (1 µM) increased the expression of NCX, while SERCA2a expression was reduced. In conclusion, aconitine increased the cytosolic [Ca2+]i by accelerating ICa-L and changing the expression of NCX and SERCA2a. Then, the elevation of cytosolic [Ca2+]i induced triggered activities and delayed after-depolarizations. Arrhythmogenesis toxicity of aconitine is related to intracellular Ca2+ signals.
Keywords: Aconitine, arrhythmias, L-type Ca2+ current, caffeine-induced Ca2+ release, NCX, SERCA2a
Introduction
A toxin produced by the aconitum plant, aconitine has been used in the past as a drug for antipyretic, analgesic and antirheumatic treatments, as well as a neurotransmission inhibitor, in China and other countries [1-4]. Yet, aconitine is known to injure both the heart and central nervous system [5-7] and its toxicities are lethal. The arrhythmogenic effects of aconitine include the induction of ventricular tachycardia (VT), torsades de pointes and ventricular fibrillation (VF) [8]. Aconitine can interact with voltage-dependent Na+ channels and suppress the conformational change of Na+ channels from the active state to the inactive state so that the membrane remains depolarized [1,9,10]. A large quantity of Na+ influx into the cytosol eventually induces triggered activities [11,12]. However, in the clinic, Ca2+ channel antagonists, such as verapamil, have been observed to have better therapeutic effects on aconitine-induced VTs rather than Na+ channel antagonists, such as quinidine [13,14]. To better understand the cardiac toxicities of aconitine, it is of paramount importance to explore the underlying mechanisms of the arrhythmogenic effects caused by aconitine.
In cardiac myocytes, L-type Ca2+ currents (ICa-L) are activated upon membrane depolarization, and Ca2+ influx through ICa-L triggers the release of Ca2+ via Ca2+ release channels (ryanodine receptors) of the sarcoplasmic reticulum [15,16]. It was reported that arrhythmias induced by aconitine had a direct relationship with the activation of RyR2 by disrupting intracellular Ca2+ homeostasis [17]. However, in neonatal rat ventricular myocytes, aconitine decreased the density of ICa-L [18]. The inhibition of ICa-L resulting in retrograde communication between the SR Ca2+ release channel and ICa-L and the direct effect of aconitine on ICa-L are unclear.
The present study was therefore designed to elucidate the role of intracellular Ca2+ signals on aconitine-induced arrhythmias. We report here that aconitine increased intracellular [Ca2+]i by accelerating ICa-L and changing the expression of NCX and SERCA2a and induced triggered activities and delayed after-depolarizations (DADs) in freshly isolated rat ventricular myocytes.
Materials and methods
Animals
Pathogen-free Sprague-Dawley male adult rats weighing 200-250 g (Grade Ⅱ, certificate No.09-2-1) were purchased from the Experimental Animal Center of Harbin Medical University. The procedures for handling the animals were in accordance with the guidelines set by the Ethics Committee of Harbin Medical University.
Cell isolation
Ventricular myocytes were isolated by an enzymatic dissociation method similar to that described previously [19,20]. Rats were anaesthetized with pentobarbital by peritoneal injection. The heart was removed immediately and mounted via the aorta to the Langendorff perfusion system. The heart was initially perfused with Ca2+-containing Tyrode solution containing the following (in mM: NaCl 126, KCl 5.4, MgCl2 1, CaCl2 1.8, NaH2PO4 0.33, glucose 10, and HEPES 10, pH adjusted with NaOH to 7.4). After the effluent was clear of blood, the heart was perfused with Ca2+-free Tyrode solution (omitting CaCl2 from the Tyrode solution) for approximately 20 min, followed by perfusion with the same Ca2+-free solution containing 0.05% collagenase and 0.1% bovine serum albumin. All of the perfusates were bubbled with 95% O2 + 5% CO2. The dispersed cells were stored in KB medium (in mM: glutamic acid 70, taurine 15, KCl 30, KH2PO4 10, MgCl2 0.5, egtazic acid 0.5, HEPES 10, glucose 10, and 1% albumin, pH adjusted with KOH to 7.4). After one hour, only Ca2+-tolerant, clearly striated, rod-shaped cells without any blebs were studied.
Patch-clamp techniques
The patch-clamp recording techniques used have been previously described in detail [21,22]. Calcium currents were recorded using standard whole-cell patch clamp techniques and voltage-clamp experiments were performed with an Axopatch 700A amplifier (Axon Instruments). The recording electrodes (Borosilicate glass, Sutter) were pulled (P-87, Sutter Instruments) and polished (F-83, Narishige) down to 2-3 MΩ when filled with pipette solution (in mM: NaCl 10, KCl 50, K2SO4 50, and MgCl2 5.0, pH adjusted with NaOH to 7.35). After the formation of the gigaohm-seal, the capacitance was electronically compensated and the cell membrane under the pipette tip was then ruptured by a brief increase in suction, forming the whole-cell recording configuration. After 2-5 min period for intracellular dialysis, the Tyrode solution was changed by bath perfusion of extracellular recording solution designed for Ca2+ current recordings (in mM: Tris-HCl 136, CsCl 5.4, MgCl2 1, CaCl2 1.8, NaH2PO4 0.33, glucose 10, and HEPES 10, pH adjusted with Tris-OH to 7.4). All cells were recorded at room temperature (22 - 23℃). Current amplitude data of each cell were normalized to its cell capacitance (current density, pA/pF). Current voltage relationship (I-V curve) was presented by the currents normalized by the peak currents. Voltage-dependent activation and steady-state inactivation profiles were calculated by Boltzmann fitting function. The activation curves were obtained by fitting the data points to a Boltzmann equation of the form GCa/GCamax =1/{ 1 + exp[(Vh - Vm) / k] } where Vh and k represent the voltage of activation midpoint and slope factor, respectively where Gmax is the maximum conductance of the voltage-gated Ca2+ channels, Vrev is the extrapolated reversal potential of ICa, V½ is the potential for half - maximal conductance, and k is the slope. The curve for voltage dependence of steady state ICa inactivation was obtained by fitting the data to a Boltzmann distribution of the form : I / Imax = I / { I + exp[( Vm - Vh ) / k] }, where I represents the current amplitude; Imax, its maximum; Vm, the potential of pre-pulse; Vh, the half-maximal inactivation potential; and k, the slope factor.
The speed of ICa-L recovery from inactivation was assessed using the following protocol: a 20-ms pre-pulse (Ip) was followed by a variable recovery period and a 20-ms test pulse (It) to assess the amount of current recovered. Each two-pulse sequence was separated by a 50-s interval.
Measurement of intracellular [Ca2+]i
The measurements of the intracellular [Ca2+]i were described previously [20]. The isolated cardiomyocytes adhered to the bottom of self-made chambers for 1 hour and then were loaded in a 10 μM Fluo 3-AM working solution at 37°C for 40-60 min, and washed with the Tyrode solution containing 1.8 mM CaCl2 to remove the extracellular Fluo 3-AM. The fluorescence intensity (FI) was detected by confocal microscopy (Insight Plus-IQ, Meridian, USA) using a 488-nm blue laser for excitation and 530-nm laser for emission during one experimental process, 25 scans with 10-s intervals were used.
Caffeine (10 mM) was used to induce the release of Ca2+ via RyRs, and [Ca2+]i was monitored. The experimental solution contained free Ca2+ and 5 mM EGTA. The amplitude of the Ca2+ transient was used as a measure of the SR Ca2+ content by evaluating F/F0. Aconitine (1 µM) was used as a pretreatment for 10 min before the administration of caffeine.
Cardiac myocyte cultures
Primary cultures of neonatal rat cardiac ventricular myocytes were performed as described previously [20]. In brief, hearts from 1-2-day-old SD rats were placed in an ice-cold 1× phosphate-buffered saline solution. After repeated rinsing, the ventricles were minced with scissors. The minced tissue and ventricular cells were dispersed by digestion with collagenase type IV (0.45 mg/ml), 0.1% trypsin, and 15 μg/ml DNase I. The cardiomyocytes (0.33 ×106 cells/ml) were cultured in cardiac myocyte culture medium containing Dulbecco's modified Eagle's medium/F-12 supplemented with 5% horse serum, 4 μg/ml of transferrin, 0.7 ng/ml of sodium selenite, 2 g/liter of bovine serum albumin, 3 mmol/liter of pyruvic acid, 15 mmol/liter of HEPES, 100 μmol/liter of ascorbic acid, 100 μg/ml of ampicillin, 5 μg/ml of linoleic acid, 1% penicillin, 1% streptomycin, and 100 μmol/liter of 5-bromo-2'-deoxyuridine and were seeded into six-well plates.
Western blot analysis
Total protein samples were extracted from the cultured neonatal rat cardiac ventricular myocytes including the control group and the group pretreated with aconitine for 48 h. The sample mixture was homogenized mechanically, incubated for 20 min on ice, and then centrifuged at 100×g for 10 min to remove the debris and nuclei. The supernatant was then collected, and the pellet was homogenized again. The latter procedure was repeated three times to increase the efficiency of the protein extraction. The collected supernatant was finally centrifuged at 100,000×g using an Optima LE-80K ultracentrifuge (Beckman, Instruments, CA, USA). The pellet corresponding to the membrane fraction was resuspended in 1% Triton-X100 cold extraction buffer and stored at -80ºC. Membrane proteins (~50 µg) were fractionated by SDS polyacrylamide gel electrophoresis (7.5% polyacrylamide gels) and transferred to polyvinyl difluoride (PVDF) membranes. After the transfer, the membranes were blotted overnight with anti-NCX (1:1000) antibodies (Santa Cruz Biotechnology, CA, USA) and anti-SERCA2a (Santa Cruz Biotechnology, CA, USA). The next day, the membranes were incubated for 2 h with the secondary antibody (goat anti-rabbit IgG, 1:10000). Bands were visualized with enhanced chemiluminescence. GAPDH was used as an internal loading control for the protein samples using an anti-GAPDH antibody. The Western blot bands were quantified using Quantity One software by measuring the band intensity (Area x OD) for each group and normalizing to GAPDH. The final results are expressed as fold changes by normalizing the data to the control values.
Statistical analysis
The data are presented as the means ± S.E.M and the paired Student's t-test was used as appropriate to evaluate the statistical significance of the differences between the two group means. Values of two-tailed p <0.05 were considered a statistically significant difference.
Results
Effect of aconitine on action potential duration (APD) of rat ventricular myocytes
Action potential values were recorded under the current-clamp mode, and a stimulatory voltage that was sufficient to induce an action potential was used in this experiment. Fig. 1A shows that the APD for both 50% and 90% repolarization (APD50 and APD90) were prolonged significantly by 1 µM of aconitine. When the aconitine concentration was elevated to 5 µM, we observed triggered activities and DADs (6 of 8 cases, Fig. 1B). Verapamil (10 µM) showed a marked reversal effect. However, Quinidine (10 µM) failed to reverse the aconitine-induced prolongation of the APD. The detailed results are listed in Table 1.
Effect of aconitine on APD in rat ventricular myocytes. A. a) Representative recordings of APD. b) Effect of aconitine on APD50 and APD90. B. DADs and triggered activity were observed following 5 µM and 10 µM aconitine administration. DADs were not abolished by washout. a) control; b)1 µM aconitine; c) 5 µM aconitine; d) 10 µM aconitine.
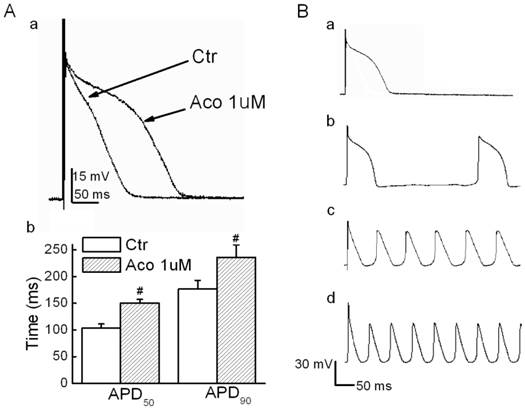
Effects of aconitine and antiarrhythmic drugs on the APD in rat ventricular myocytes.
| Group | RP(mV) | OS(mV) | APD50 (ms) | APD90(ms) |
|---|---|---|---|---|
| Ctr | -72.5±5.98 | 43.2±3.59 | 105.4±7.71 | 154.2±7.02 |
| Aco 1 µM | -70.5±3.44 | 46.7±2.34 | 178.4±16.43 ** | 2376.0±23.22 ** |
| Aco1 µM +Qui | -64.8±9.71 | 29.9±9.31 | 208.8±83.38** | 316.15±67.39**a |
| Aco1 µM +Ver | -72.4±2.68 | 32.4±5.09 | 105.6±10.81a | 147.7±32.38a |
*p<0.05 vs control **p<0.01 vs control ap<0.01 vs aconitine (RP: resting potential; OS: overshoot)
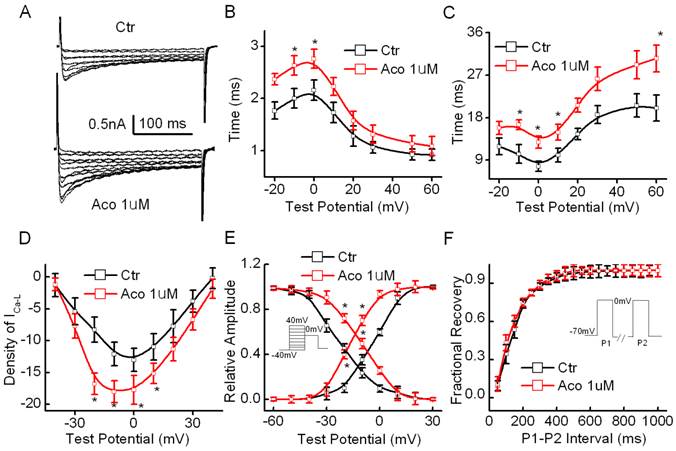
Effect of aconitine on the current-voltage relationships of ICa-L
The cell capacitance of the cardiomyocytes used in our experiments was measured as 71.3 ± 18.6 pF, and the series resistance (Rs) was 5.32 ± 1.06 MΩ (n=20) during the recording process. The results showed that the density of ICa-L in the rat ventricular myocytes increased significantly from 12.77 ± 3.12 pA/pF to 18.98 ± 3.89 pA/pF (p<0.05, n =10) after exposure to 1 µM aconitine. Fig. 2B shows representative recordings of ICa-L and an I-V curve of ICa-L. Fig. 2C and 2D show that aconitine increased the time course of the activation and inactivation of ICa-L. Fig. 2E and 2F show the effect of aconitine on the kinetics of ICa-L. V1/2 of the activation curve was altered from -3.64±0.81 mV to -16.7 ± 0.55 mV (p<0.05, n=8) by aconitine. The inactivation curve shifted markedly toward the right by 1 µM aconitine. V1/2 changed from -22.7 ± 1.19 mV to -9.6 ± 0.71 mV (p<0.05, n=8).
Effect of aconitine on ICa-L in the single cell of rat ventricular myocytes. A. Representative curves recorded before and after 1 µM aconitine was perfused. Currents were elicited by 300-ms voltage steps from -60 to +40 mV from a holding potential of -70 mV at an inter-pulse interval of 10 s. B. Effect of aconitine on the time course of activation of ICa-L. C. Effect of aconitine on the time course of inactivation of ICa-L. D. Effect of aconitine on the voltage-current density relationship curve of ICa-L. E. Changes of activation and inactivation of ICa-L under exposure to 1 µM aconitine. F. Effect of 1 µM aconitine on the recovery of Ca2+ current from inactivation. Raw data were fitted by a mono-exponential function.
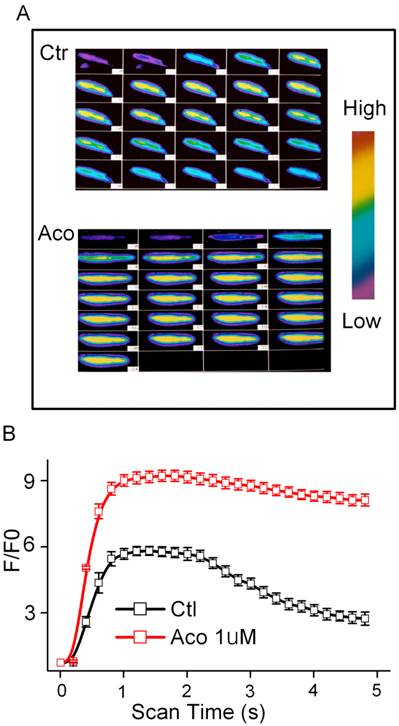
Effects of aconitine on intracellular [Ca2+]i
Aconitine (1 μM) has no effect on the resting level of FI in the presence or absence of extracellular Ca2+ (data not shown). The Ca2+ content released from the SR during the caffeine contracture was measured as FI. Superimposed Ca2+ transients were performed in the control and incubation of 1 μM aconitine groups (Fig. 3). It was shown that Ca2+ transients markedly increased, whereas the reverse process was slower than normal (Fig. 3). The peak of FI /F0 of [Ca2+]i increased from 4.74 ± 0.27 to 9.15 ± 0.36 by aconitine treatment (p<0.05, n=10).
Effect of aconitine on intracellular [Ca2+]i in single ventricular myocytes from rat. A. Representative fluorescent images of ventricular myocytes loaded with Fluo-3/AM under control condition and 1 µM aconitine. Left: cytosolic [Ca2+]i induced by 60 mM KCl; Right: cytosolic [Ca2+]i induced by 300 μM caffeine. B. Changes in [Ca2+]i induced by KCl is represented by fluorescence intensity. C. Changes in [Ca2+]i induced by caffeine is represented by fluorescence intensity.
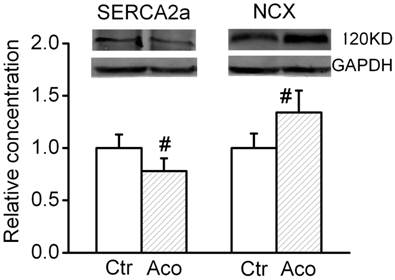
Expression level of SERCA2a and Na+/Ca2+ exchanger (NCX)
SERCA2a is a Ca2+ ATPase that transfers Ca2+ from the cytosol of the cell to the lumen of the SR at the expense of ATP hydrolysis during muscle relaxation. NCX is an antiporter membrane protein that removes Ca2+ from cells. It uses the energy that is stored in the electrochemical gradient of Na+ by allowing Na+ to flow down its gradient across the plasma membrane in exchange for the countertransport of Ca2+. We further investigated possible changes in the expression levels of cytosolic Ca2+ handling proteins. As shown in Fig. 4, after treatment with 1 µM aconitine, the SERCA2a expression significant decreased and NCX expression increased.
Effect of aconitine on expression of SERCA2a and NCX.
Discussion
In this study, we found that in freshly-isolated rat ventricular myocytes, aconitine increased intracellular [Ca2+]i by affecting the Ca2+ signals and induced triggered activities and DADs, which may contribute to its arrhythmogenic effect. This conclusion was mainly based on the observations that (1) aconitine prolonged the APD and at higher concentrations, induced triggered activities and DADs; (2) aconitine increased ICa-L by accelerating activation and delaying inactivation; (3) caffeine-induced Ca2+ release was markedly increased by aconitine; (4) aconitine reduced the expression level of SERCA2a and increased the expression level of NCX. Therefore, the present study provides insight into a useful method for preventing aconitine-induced arrhythmias by blocking ICa-L in the clinic.
In our experiment, we found that 1 µM aconitine significantly prolonged APD50 and APD90 in isolated rat ventricular myocytes. When its concentration was elevated to 5 µM and 10 µM, aconitine induced triggered activities and DADs (as shown in Fig. 1B). It is well known that aconitine can induce serious arrhythmias by suppressing Na+ channels. However, in our experiment, quinidine (10 µM) failed to suppress DADs and triggered activities induced by aconitine. In Table 1, verapamil shortened the aconitine-induced prolongation of APD and suppressed DADs and triggered activities. It is well-known that DADs are triggered by Ca2+ overload caused by enhanced Ca2+ entry and membrane depolarization in response to non-electrically driven (i.e., spontaneous) Ca2+ release from the SR [23]. In our experiment, we found aconitine prolonged APD and in the meanwhile depolarized the membrane potential (as Table 1 shown), so the membrane potential was prone to be excited and triggered the membrane oscillations. These results further suggested that aconitine-induced triggered activities were related to intracellular Ca2+ signals.
In Fig. 2, aconitine accelerated the activation of ICa-L and delayed the inactivation of ICa-L. So aconitine increased Ca2+ influx through ICa-L. On the contrary, Fu et al. reported that ICa-L was inhibited in cultured neonatal rat ventricular myocytes [17]. These inconsistent data may result from the difference in the two types of myocytes. Ionic channel characters of neonatal and adult ventricular myocytes are not completely identical. ICa-L regulates action potential repolarization and plays an important role in the development of cardiac arrhythmias such as ventricular fibrillation, the leading cause of sudden cardiac death [23,24]. ICa-L is activated upon membrane depolarization and Ca2+ influx through ICa-L triggers the release of Ca2+ via the Ca2+ release channels (ryanodine receptors) of the sarcoplasmic reticulum. During normal AP, early ICa-L peaks trigger robust SR release before partially inactivating due to two processes, Ca2+-dependent inactivation and voltage-dependent inactivation. Aconitine delayed the rate and degree of inactivation of ICa-L which may affect the intracellular Ca2+ cycling dynamic process.
So we investigated SR Ca2+ release by caffeine stimulation. The rapid application of caffeine induces a [Ca2+]i transient, and the amplitude can be used as an index of the SR Ca2+ releasable content [5]. Fig. 3 demonstrates that aconitine increased caffeine-induced Ca2+ release in cardiomyocytes. The enhanced SR Ca2+ release and Ca2+ entry via ICa-L led to an increase in the intracellular [Ca2+]i. These data were consistent with that reported with cultured neonatal rat ventricular myocytes showing that the level of caffeine-induced Ca2+ release was increased [17]. In normal conditions for relaxation to occur, [Ca2+]i must decline allowing Ca2+ to be transported out of the cytosol mainly via the sarcolemmal Na+/Ca2+ exchange (NCX) and the SR Ca2+-ATPase (SERCA), which takes Ca2+ back into the SR. In our study, we found in cultured neonatal myocytes with pretreatment of aconitine the expression level of SERCA decreased, whereas the expression level of NCX increased. NCX can operate in both the Ca2+ efflux and influx (or reverse) modes, depending on the internal and external concentration of both Na+ and Ca2+. In myocytes induced by aconitine, [Na+]i was elevated, and NCX works almost exclusively in the Na+ extrusion mode. But our experiment has not directly demonstrated NCX mode. The amount of Ca2+ influx through NCX can increase greatly. This influx will raise the cellular and SR Ca2+ content, resulting in larger Ca2+ transients.
Under the above-mentioned condition, intracellular Ca2+ markedly increased and induced Ca2+ overload, which increased the frequency of propagating Ca2+ waves, and this increase escalates the propensity for triggered arrhythmia [25,26]. Intracellular Ca2+ overload triggered by Ca2+ signals may underlie a mechanism of aconitine-induced cardiac arrhythmia. The results were consistent with that reported aconitine induced high frequency intracellular Ca2+ oscillation and typical Ca2+ sparks disappeared in cultured cardiomyocytes [27]. It was reported that in aconitine-induced arrhythmia, intravenous injection of Ca2+ preparations promoted recovery of stable sinus rhythm and decreased animal mortality [28]. The Na+ and Ca2+ intracellular imbalance were related with aconitine-induced cardiac arrhythmias.
Gong et al. reported that aconitine inhibited transient K+ current in ventricular myocytes [29]. It was reported that aconitine blocked ultra-rapid delayed rectifier K+ currents in H9c2 myoblasts [30] and HERG and Kv1.5 channels expressed in Xenopus laevis oocytes [31]. Inhibition of K+ currents could lead to excessive prolongation of APD and disturb the electrical character of the membrane so that myocytes are liable to induce DADs and triggered activities.
In our experiment, aconitine increased intracellular [Ca2+]i by accelerating ICa-L, leading to triggered activities and DADs, which may contribute to its arrhythmogenic toxicity.
Conclusion
The present study was designed to elucidate the role of intracellular Ca2+ signals on aconitine-induced arrhythmias. We reported here that aconitine prolonged APD and at higher concentration induced triggered activities and delayed after-depolarizations (DADs) in freshly isolated rat ventricular myocytes, The mechanisms was related with the elevation of intracellular [Ca2+]i by accelerating ICa-L, increasing the expression of NCX and decreasing the expression of SERCA2a.
Competing Interests
The authors have declared that no conflict of interest exists.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No.81202525 and No.81173051) and Returned Oversea Scholars' Foundation of Heilongjiang Province (No. LC2011C04).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fraser SP, Salvador V, Manning EA, Mizal J, Altum S, Raza M. et al. Contribution of functional voltage-gated Nat channel expression to cell behavior involved in the metastatic cascade in rat prostate cancer: I. Lateral motility. J. Cell. Physiol. 2003;195:479-87
2. Sato H, Yamada C, Konno C, Ohizumi Y, Endo K, Hikino H. Pharmacological actions of aconitine alkaloids. Tohoku. J. Exp. Med. 1979;128:175-87
3. Hikino H, Konno C, Takata H, Yamada Y, Yamada C, Ohizumi Y. et al. Anti-inflammatory principles of aconitum roots. J. Pharm. Dyn. 1980;3:514-25
4. Herzog W, Feibel R, Bryant S. The effect of aconitine on the giant axon of the squid. J. Gen. Physiol. 1964;47:719-33
5. Catterall WA, Trainer V, Baden DG. Molecular properties of the sodium channel: a receptor for multipleneurotoxins. Bull. Soc. Pathol. Exot. 1992;85:481-85
6. Honerjager P, Meissner A. The positive inotropic effect of aconitine. Naunyn-Schiedeberg's. Arch. Pharmacol. 1983;322:49-58
7. Amer A. The effects of aconitum alkaloids on the central nervous system. Prog. Neurobiol. 1998;56:211-35
8. Lu HR, De Clerck F. R56865, a Na+/Ca(2+)-overload inhibitor, protects against aconitine-induced cardiac arrhythmias in vivo. J. Cardiovasc. Pharmacol. 1993;22:120-25
9. Wright SN. Comparison of aconitine-modified human heart (hH1) and rat skeletal (m1) muscle Nat channels: An important role for external Nat ions. J. Physiol. 2002;538:759-71
10. Wang SY, Wang GK. Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cell. Signal. 2003;15:151-9
11. Sawanobori T, Hirano Y, Hiraoka M. Aconitine-induced delayed after depolarization in frog atrium and guinea-pig papillary muscles in the presence of low concentration of calcium. Jpn. J. Physiol. 1987;37:59-79
12. Watano T, Harada Y, Harada K, Nishimura N. Effect of Na+/Ca2+ exchange inhibitor, KB-R7943 on ouabain-induced arrhythmias in guinea-pigs. Br. J. Pharmacol. 1999;127:1846-50
13. Van Landeghem AA, De Letter EA, Lambert WE, Van Peteghem CH, Piette MH. Aconitine involvement in an unusual homicide case. Int. J. Legal. Med. 2007;121:214-19
14. Takahara A, Uneyama H, Sasaki N, Ueda H, Dohmoto H, Shoji M. et al. Effects of AH-1058, a new antiarrhythmic drug, on experimental arrhythmias and cardiac membrane currents. J. cardiovasc. Pharm. 1999;33:625-32
15. Eisner DA, Kashimura T, Venetucci LA, Trafford AW. From the ryanodine receptor to cardiac arrhythmias. Circ. J. 2009;73:1561-67
16. Bito V, Heinzel FR, Biesmans L, Antoons G, Sipido KR. Crosstalk between L-type Ca2+ channels and the sarcoplasmic reticulum: alterations during cardiac remodelling. Cardiovasc. Res. 2008;77:315-24
17. Fu M, Wu M, Wang JF, Qiao YJ, Wang Z. Disruption of the intracellular Ca2+ homeostasis in the cardiac excitation contraction coupling is a crucial mechanism of arrhythmic toxicity in aconitine-induced cardiomyocytes. Biochem. Biophys. Res. Commun. 2007;354:929-36
18. Fu M, Li RX, Fan L, He GW, Thornburg KL, Wang Z. Sarcoplasmic reticulum Ca2+ release channel ryanodine receptor (RyR2) plays a crucial role in aconitine-induced arrhythmias. Biochem. Pharmacol. 2008;75:2147-56
19. Liu Y, Sun L, Pan Z, Bai Y, Wang N, Zhao J. et al. Overexpression of m(3) muscarinic receptor is a novel strategy for preventing sudden cardiac death in transgenic mice. Mol. Med. 2011;17:1179-87
20. Cai B, Gong D, Chen N, Li J, Wang G, Lu Y. et al. The negative inotropic effects of homocysteine were prevented by matrine via the regulating intracellular calcium level. Int. J. Cardiol. 2011;150:113-15
21. Chen X, Shan H, Zhao J, Hong Y, Bai Y, Sun I. et al. L-type calcium current (ICa,L) and inward rectifier potassium current (IK1) are involved in QT prolongation induced by arsenic trioxide in rat. Cell. Physiol. Biochem. 2010;26:967-74
22. Zhou Y, Xu W, Han R, Zhou J, Pan Z, Rong H. et al. Matrine inhibits pacing induced atrial fibrillation by modulating I(KM3) and I(Ca-L). Int. J. Biol. Sci. 2012;8:150-8
23. Moroz VM, Lipnitskii TN. Dysfunction of ionic channels in cardiomyocyte sarcolemma and cardiac arrhythmias. Bull. Exp. Biol. Med. 2006;141:397-9
24. Kranias EG, Bers DM. Calcium and cardiomyopathies. Subcell. Biochem. 2007;45:523-37
25. Wang YJ, Chen BS, Lin MW, Lin AA, Peng H, Sung RJ. et al. Time-dependent block of ultra rapid-delayed rectifier K+ currents by aconitine, a potent cardiotoxin, in heart-derived H9c2 myoblasts and in neonatal rat ventricular myocytes. Toxicol. Sci. 2008;106:454-63
26. Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F. et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001;10:189-94
27. Zhang SW, Liu Y, Huang GZ, Liu L. Aconitine alters connexin43 phosphorylation status and [Ca2+] oscillation patterns in cultured ventricular myocytes of neonatal rats. Toxicol In Vitro. 2007;21(8):1476-85
28. Moroz VM, Lipnitskii TN. Dysfunction of ionic channels in cardiomyocyte sarcolemma and cardiac arrhythmias. Bull Exp Biol Med. 2006;141(4):397-9
29. Gong DM, Shan HL, Zhou YH, Dong DL, Yang BF. The ion targets of arrhythmias induced by ouabain and aconitine in guinea pig and rat ventricular myocytes. Yaoxuexuebao. 2004;39:328-32
30. Jiang D, Xiao B, Zhang L, Chen SR. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ. Res. 2002;91:218-25
31. Li Y, Tu D, Xiao H, Du Y, Liao Y, Dong S. Aconitine blocks HERG and Kv1.5 potassium channels. J. Ethnopharmacol. 2010;131:187-95
Author Biology
Dr Yuhong Zhou works for the Department of Pharmacology, Harbin Medical University, which focuses on understanding the mechanism of cardiovascular drug. The Department is the State-Province Key Laboratories of Biomedicine-Pharmaceutics of China and one of the Key Laboratory of Cardiovascular Research affiliated to Ministry of Education in China. Dr Zhou is an associated professor and research scientist with awards for outstanding and innovative research in the molecular and electrophysiological mechanisms of Chinese traditional herbs.
Dr Hongli Shan is a research scientist with 15 years of research experience in the cardiovascular Pharmacology. Dr. Shan's research has been fully funded by the funding agencies such as the National Nature & Science Foundation, Provincial Key Research Foundation, Provincial Educational Committee and highly horned for several time by central and local department of science and technology. She has published more than 45 research papers internationally.
Author contact
![]() Corresponding author: Dr. Hong-li Shan, Department of Pharmacology, Harbin Medical University, Bao-Jian Road 157, Harbin 150081, E-mail address: shanhonglicom.cn
Corresponding author: Dr. Hong-li Shan, Department of Pharmacology, Harbin Medical University, Bao-Jian Road 157, Harbin 150081, E-mail address: shanhonglicom.cn