Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(5):634-640. doi:10.7150/ijms.5547 This issue Cite
Research Paper
Proliferation Enhanced By NGF-NTRK1 Signaling Makes Pancreatic Cancer Cells More Sensitive To 2DG-Induced Apoptosis
Yao Cheng, Dong-mei Diao, Hao Zhang, Yong-Chun Song, Cheng-Xue Dang ![]()
The Department of Surgical Oncology the First Affiliated Hospital, Xi'an Jiaotong University College of Medicine, 277 W. Yanta Road, Xi'an, Shaanxi 710061, China
Received 2012-11-14; Accepted 2013-3-20; Published 2013-3-23
Abstract
Rapidly proliferating cancer cells rely on increased glucose consumption for survival. The glucose analog 2-deoxy-D-glucose (2DG) cannot complete glycolysis and inhibits the growth of many types of cancers. It is unknown whether reduced glycolysis inhibits the growth of pancreatic cancer. Activation of nerve growth factor (NGF)-neurotrophic tyrosine kinase receptor type 1 (NTRK1) signaling leads to enhanced proliferation of these cells. We investigated the effect of 2DG treatment on the viability of NTRK1-transfected pancreatic cancer cells. After treatment with 2DG, the viability of pancreatic cancer cells was evaluated by MTT assay. SB203580 (a specific inhibitor of the p38-MAPK pathway) and PD98059 (an MAP2K1 [mitogen-activated protein kinase kinase 1, previously, MEK1] inhibitor) were used to inhibit p38-MAPK and ERKs, respectively. The percentage of apoptotic cells was determined by flow cytometry. Overexpression of NTRK1 in pancreatic cancer cells resulted in increased cell proliferation, which was reduced by PD98059-mediated inhibition of ERKs but not by suppression of p38-MAPK with SB203580. After treatment with 2DG, the percentage of apoptotic cells was greater in those with high expression of NTRK1 than in cells with low NTRK1 expression. Blocking the p38-MAPK pathway with SB203580 effectively abolished the apoptosis induced by 2DG. We conclude that pancreatic cancer cells with a high expression of NTRK1 are more sensitive to 2DG-induced apoptosis, through the p38-MAPK pathway.
Keywords: NGF, NTRK1, pancreatic cancer, glycolysis, 2DG
Introduction
Nerve growth factor (NGF) is a member of the well-studied neurotrophin family, proteins with important roles in the regulation of cell growth, proliferation, survival, and death [1, 2]. NGF and its high affinity receptor, neurotrophic tyrosine kinase receptor type 1 (NTRK1), promote cell survival, proliferation, and resistance to cancer chemotherapy by activating the extracellular-signal-regulated kinase (ERK), phosphatidylinositide 3-kinase (PI3K)/ AKT1, and p38 mitogen-activated protein kinase (MAPK) signaling pathways [3-7]. However, in some cell types NGF induces cell differentiation or apoptosis, via p53[8] or c-Jun N-terminal kinases (JNKs) [9], respectively.
High levels of NGF and NTRK1 in pancreatic cancers and the perineurium of pancreatic nerves are closely correlated with perineural invasion [10], tumor aggressiveness, and unfavorable prognosis, and are accompanied by activation of both ERKs and p38-MAPK signaling. The function of the activation of p38-MAPK signaling by NGF is not well understood. It has been shown that activation of p38-MAPK is associated with proliferation, migration, and resistance to apoptosis [11, 12]. Yet p38-MAPK also activates p53, induces cell-cycle arrest, apoptosis, and differentiation, and makes cells sensitive to chemotherapy [9, 13-17].
In previous studies [18] we demonstrated increased cell proliferation in NTRK1-transfected human pancreatic cancer cells, while knockdown of NTRK1 by small interfering RNA (siRNA) inhibited cell proliferation and sensitized pancreatic cancer to the chemotherapy drug gemcitabine. These results support targeting the NGF-NTRK1 pathway in pancreatic cancer therapy. However, the NTRK inhibitor CEP-701 (lestaurtinib) demonstrated significant damages to other tissues with high expression of NTRK1. Thus, it is important to develop an alternative strategy for the treatment of pancreatic cancers with increased levels of NGF and NTRK1[19].
Unlike normal cells that gain energy through oxidative phosphorylation in mitochondria, the increased proliferation of cells in most types of cancers depends upon accelerated anaerobic glycolysis in the cytosol [20-22], a phenomenon known as the Warburg effect [23].
Therefore, targeting the anaerobic glycolytic pathway may preferentially kill malignant cells while sparing the normal [24, 25]. Since pancreatic cancer cells have been shown to have an increased utilization of glucose [26, 27], it is intriguing to target the glycolytic metabolic pathway for the treatment of pancreatic cancers [28].
To test if inhibition of glycolysis of pancreatic cancer cells in vitro results in reduced cell proliferation, we evaluated cell viability after glucose deprivation induced by the nonmetabolizable glucose analog 2-deoxy-D-glucose (2DG) in NTRK1-transfected ASPC-1, BXPC-3, and Panc-1 pancreatic cancer cells.
Materials and Methods
Reagents
Dimethyl sulfoxide (DMSO), 2DG, G418 sulfate, β-NGF, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (USA). Radioimmunoprecipitation (RIPA) buffer and phenylmethylsulfonyl fluoride (PMSF) were obtained from Beyotime (China). Anti- NTRK1, Anti-p-p38, ERK inhibitor PD98059, and p38 inhibitor SB203580 were from Abcam (USA). Anti-β-actin antibody and secondary antibody were from Abnova.
Cell culture
Cells of the human pancreatic cancer cell lines ASPC-1, BXPC-3, and Panc-1 were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). ASPC-1 cells were grown in RPMI-1640 and BXPC-3 or Panc-1 cells in Dulbecco's modified Eagle's medium (DMEM; both from Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone), penicillin 100 U/mL, and streptomycin 100 μg/mL (Gibco). The cultures were maintained at 37 °C in a 5% CO2 incubator.
Establishment of stable transfected cell lines
The plasmid pcDNA3.1-NTRK1 was provided by Wei Liu (Fourth Military Medical University, Xi'an, China). All transfections were performed using X-tremeGENE HP in accordance with the manufacturer's instructions, at the ratio of 1:1 (Roche). For generation of NTRK1-transfected or non-NTRK1-transfected cell lines, ASPC-1 cells were transfected with pcDNA3.1-NTRK1 vector or pcDNA3.1 blank vector, respectively. Cells were maintained in 200 ng/mL G418 for 21 days. Individual clones were screened for expression of NTRK1 by Western blot analysis.
Transfection of siRNA
Human NTRK1-specific and control siRNAs were purchased from Biocompare (SMARTpool, Charlottesville, VA). BXPC-3 or Panc-1 cells were plated in 6-well plates and incubated for 24 h prior to transfection. Eight microliters of siPORT Amine transfection reagent (Ambion) were added to 200 μL serum-free medium and incubated at room temperature for 15 min. The transfection reagent/siRNA complexes were added to each well containing 800 μL medium with 10% FBS. Six hours later, 1 mL DMEM containing 10% FBS was added into each well.
MTT assay of cell growth inhibition
Cells were seeded (3000/well) in 96-well plates for 24 hours. 2DG, PD98059, or SB203580 was added and incubated for the indicated time at 37 °C. MTT (0.5 mg/mL in PBS) was added to each well and incubated for 4 h at 37 °C. The media was then discarded and 100 μL DMSO was added. After agitation for 10 min on a shaker, absorbance was read at 550 nm on a spectrophotometer. Data were normalized relative to the untreated group.
Western blot
Cells were lysed on ice with modified RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) containing 25 μg/mL leupeptin, 1 mM sodium orthovanadate, 2 mM ethylenediaminetetraacetic acid (EDTA), and 1 mM PMSF. The concentration of protein was determined using the bicinchoninic acid assay (Beyotime). Twenty micrograms of total protein were loaded onto an 8% SDS-polyacrylamide gel, electrophoresed, and blotted onto a polyvinylidene difluoride (PVDF) membrane. Proteins were probed with the primary antibody overnight at 4 °C and the secondary antibody at room temperature for 1 hr. Immunoreactivity was detected by the electrochemiluminescence (ECL) system and normalized to β-actin.
Detection of apoptosis
Twenty-four hours after treatment, 106 of cells were harvested by centrifuging at 200 ×g for 5 min. Cells were resuspended in 100 μL of Annexin-V-FLUOS (Roche), incubated at room temperature for 15 min, and subjected to flow cytometry.
Statistical analysis
All experiments were repeated in triplicate. Data were analyzed by SPSS13.0 software using Student's t-test. Probability values (P) < 0.05 were considered statistically significant.
Results
NGF-NTRK1 pathway enhances pancreatic cancer cell proliferation depends on ERKs
NGF-NTRK1 signaling enhances cell proliferation independently on the p38-MAPK pathway. NGF promotes proliferation, invasion and survival of many types of cells with high expression of NTRK1 including pancreatic cancer cells. In NTRK1-transfected ASPC-1 cells, 200 ng/mL NGF for 72 h increased the cell number by 1.5-fold in comparison to untreated group (P < 0.05). In contrast, NGF showed no obvious influence on the growth of ASPC-1 cells transfected with the empty vector (Fig. 1A). Thus the growth-stimulatory effects of NGF depend on the availability of its receptor NTRK1. Similar results were observed in BXPC-3 and Panc-1 cells (Fig. 1B and C).
NTRK1 signaling enhances cell proliferation of pancreatic cancer cells. ASPC-1 (A), Panc-1 (B) and BXPC-3 (C) cells were treated as indicated. MTT was applied to determine cell viability spectrophotometrically at 24 h, 48 h, and 72 h following treatment. (D). Western blot analysis p-NTRK1, p-ERKs, and p-p38, with β-actin as loading control.
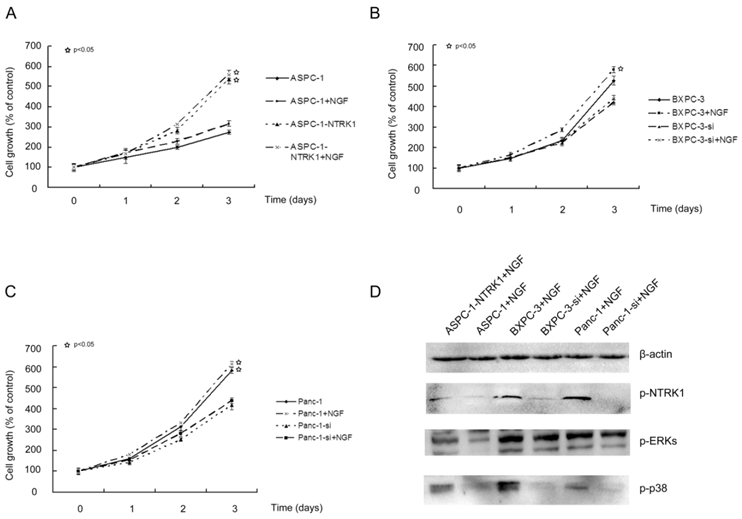
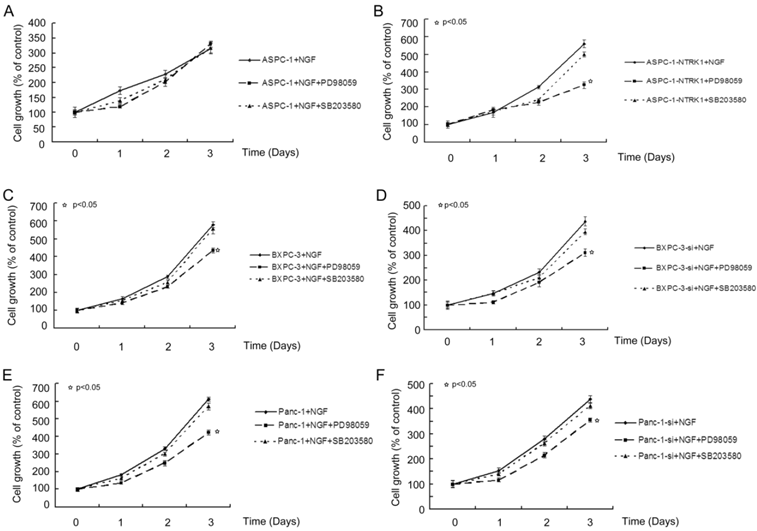
NGF-treated NTRK1-transfected cells displayed a dramatic increase in the phosphorylation of ERK and p38 (Fig. 1D). This indicates that NGF-NTRK1 pathway upregulates the p38-MAPK pathway and ERKs [29, 30]. To test whether these signaling pathways are involved in the enhanced proliferation response to NGF-NTRK1 pathway activation, we inhibited p38-MAPK with SB203580 and ERKs with PD98059. We found that, in the presence of NGF, PD98059 but not SB203580 significantly decreased the cell viability of cells transfected with NTRK1 after 72 h of incubation (Fig. 2). These data showed that the enhanced cell proliferation in response to NGF-NTRK1 activation is correlated with ERKs, and is independent of the p38-MAPK pathway.
Blocking ERKs by PD98059 greatly suppressed the proliferation of cells with overexpression of NTRK1. Results are expressed as a percentage of control cells at 8 h after plating.
2DG-induced apoptosis depends on p38-MAPK signaling but not ERKs
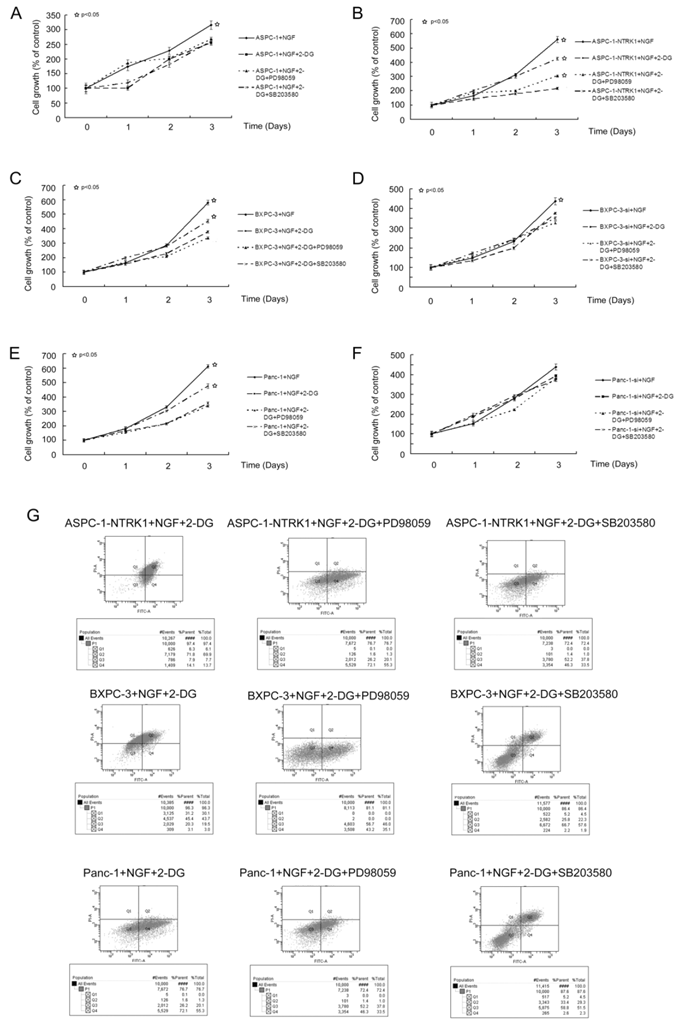
The growth of NTRK1-transfected ASPC-1 cells treated with 15 mM 2DG was 40% that of non-treated NTRK1-transfected ASPC-1 cells, in a time-dependent association. The cell growth of non-transfected ASPC-1 cells was inhibited 17% at 72 h. BXPC-3 and Panc-1 cells in which NTRK1 expression was downregulated via siRNA were less sensitive to 2DG than cells with normal NTRK1 expression (Figure 3C-F). As increased p38-MAPK and ERK activity was observed in NTRK1-transfected cells, we next checked whether upregulation of p38-MAPK or ERKs via the NGF-NTRK1 pathway were involved in 2DG-induced cell growth. For this, we treated cells with SB203580 or PD98059, and found that SB203580 but not PD98059 significantly attenuated the number of apoptotic cells caused by 2DG in all cell types that overexpressed NTRK1 (Fig. 3A-F). Consistent with this result, blocking p38-MAPK with SB203580 greatly reduced apoptosis induced by 2DG, at 24 h (Fig. 3G). These results indicate that apoptosis induced by 2DG in pancreatic cancer cells can be reduced by inhibition of the p38-MAPK pathway.
2DG-induced apoptosis depends on p38-MAPK signaling but not ERKs. (A-F) 2DG significantly reduced the growth of six types of cells, while inhibition of ERKs did not show any obvious influence. However, by inhibiting p38-MAPK with SB203580, the growth inhibition was greatly alleviated. This phenomenon was not observed in untreated ASPC-1, BXPC-3-si and Panc-1-si cells, which had a low expression of NTRK1. (G) 2DG induced apoptosis in cells with higher expression of NTRK1 after 24 h of treatment. Treatment of PD98059 did not alleviate this phenomenon, while SB203580 did.
Discussion
NTRK1 is overexpressed in various types of tumors, suggesting that NTRK1 may be a valuable therapeutic target. Unfortunate, it was found that pan-Trk inhibitors could not discriminate among NTRK1, NTRK2, and NTRK3, and therefore have relatively poor specificity. They also inhibited other targets, such as fms-related tyrosine kinase 3 (FLT3) [31]. Thus, it remains imperative to develop specifically targeted cytotoxic drugs, especially when targeted toward the hallmarks of cancer cells. For example, it is widely accepted that malignant tumors are characterized by higher rates of glucose usage and glycolysis [32]. The higher cellular proliferation rate of these malignancies depends on elevated ATP consumption [33, 34]. Thus targeting the glycolytic metabolism of cancer cells is a promising strategy for cancer therapy.
The glucose analog 2DG acts as a competitive inhibitor of glucose metabolism, causing a depletion of cellular ATP and leading to cessation of the cell cycle and apoptosis [35]. In addition, 2DG can inhibit protein glycosylation and induce accumulation of misfolded proteins in the endoplasmic reticulum, leading to endoplasmic reticulum stress and constant apoptosis [36]. 2DG has been proposed to be an anti-cancer drug for a variety of cancer cells.
In the present study, we utilized the NGF-NTRK1 pathway to enhance proliferation of pancreatic cancer cells. We found that activation of NGF-NTRK1 signaling induced cell proliferation and progression, which is in agreement with previous studies [37-39]. Interestingly, the proliferation of cells with elevated NTRK1 expression was significantly inhibited by treatment with 2DG. Our data indicates that p38-MAKP is activated by NGF-NTRK1 signaling, but may not be required for NGF-NTRK1-enhanced cell proliferation. However p38-MAPK is required for 2DG-induced apoptosis. Similarly, enhanced proliferation due to loss of PTEN makes cells more sensitive to 2DG [33].
Our results showed that NGF-NTRK1 signaling induced pancreatic cancer cell proliferation and made cells more vulnerable to glycolysis inhibition by 2DG. As enhanced dependence on glucose glycolysis is a hallmark of cancer cells, our results suggest that targeting glucose glycolysis may be an avenue for the development of anti-cancer drugs.
Acknowledgements
We thank Dr. Wei Liu for providing the plasmids used in this study. This study was supported by the National Natural Science Foundation of China (30973489).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Biarc J, Chalkley RJ, Burlingame AL, Bradshaw RA. The induction of serine/threonine protein phosphorylations by a PDGFR/TrkA chimera in stably transfected PC12 cells. Mol Cell Proteomics. 2012;11:15-30
2. Harrington AW, St Hillaire C, Zweifel LS, Glebova NO, Philippidou P, Halegoua S. et al. Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell. 2011;146:421-34
3. Bertrand T, Kothe M, Liu J, Dupuy A, Rak A, Berne PF. et al. The crystal structures of TrkA and TrkB suggest key regions for achieving selective inhibition. J Mol Biol. 2012;423:439-53
4. Jana B, Koszykowska M, Czarzasta J. Expression of nerve growth factor and its receptors, TrkA and p75, in porcine ovaries. J Reprod Dev. 2011;57:468-74
5. Li C, Macdonald JI, Hryciw T, Meakin SO. Nerve growth factor activation of the TrkA receptor induces cell death, by macropinocytosis, in medulloblastoma Daoy cells. J Neurochem. 2010;112:882-99
6. Nikoletopoulou V, Lickert H, Frade JM, Rencurel C, Giallonardo P, Zhang L. et al. Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature. 2010;467:59-63
7. Schor NF. Neuroblastoma as a neurobiological disease. J Neurooncol. 1999;41:159-66
8. Zhang J, Yan W, Chen X. p53 is required for nerve growth factor-mediated differentiation of PC12 cells via regulation of TrkA levels. Cell Death Differ. 2006;13:2118-28
9. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326-31
10. Bapat AA, Hostetter G, Von Hoff DD, Han H. Perineural invasion and associated pain in pancreatic cancer. Nat Rev Cancer. 2011;11:695-707
11. Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H. et al. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359-67
12. Zhou X, Ferraris JD, Dmitrieva NI, Liu Y, Burg MB. MKP-1 inhibits high NaCl-induced activation of p38 but does not inhibit the activation of TonEBP/OREBP: opposite roles of p38alpha and p38delta. Proc Natl Acad Sci U S A. 2008;105:5620-5
13. Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E. et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18:6845-54
14. Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC. et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23:2469-76
15. Olson JM, Hallahan AR. p38 MAP kinase: a convergence point in cancer therapy. Trends Mol Med. 2004;10:125-9
16. Sanchez-Prieto R, Sanchez-Arevalo VJ, Servitja JM, Gutkind JS. Regulation of p73 by c-Abl through the p38 MAP kinase pathway. Oncogene. 2002;21:974-9
17. Yu W, Imoto I, Inoue J, Onda M, Emi M, Inazawa J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene. 2007;26:1178-87
18. Liu D, Zhang Y, Dang C, Ma Q, Lee W, Chen W. siRNA directed against TrkA sensitizes human pancreatic cancer cells to apoptosis induced by gemcitabine through an inactivation of PI3K/Akt-dependent pathway. Oncol Rep. 2007;18:673-7
19. Festuccia C, Muzi P, Gravina GL, Millimaggi D, Speca S, Dolo V. et al. Tyrosine kinase inhibitor CEP-701 blocks the NTRK1/NGF receptor and limits the invasive capability of prostate cancer cells in vitro. Int J Oncol. 2007;30:193-200
20. Baricordi OR, Melchiorri L, Adinolfi E, Falzoni S, Chiozzi P, Buell G. et al. Increased proliferation rate of lymphoid cells transfected with the P2X(7) ATP receptor. J Biol Chem. 1999;274:33206-8
21. Bilbao PS, Santillan G, Boland R. ATP stimulates the proliferation of MCF-7 cells through the PI3K/Akt signaling pathway. Arch Biochem Biophys. 2010;499:40-8
22. Conigrave AD, van der Weyden L, Holt L, Jiang L, Wilson P, Christopherson RI. et al. Extracellular ATP-dependent suppression of proliferation and induction of differentiation of human HL-60 leukemia cells by distinct mechanisms. Biochem Pharmacol. 2000;60:1585-91
23. Giammarioli AM, Gambardella L, Barbati C, Pietraforte D, Tinari A, Alberton M. et al. Differential effects of the glycolysis inhibitor 2-deoxy-D-glucose on the activity of pro-apoptotic agents in metastatic melanoma cells, and induction of a cytoprotective autophagic response. Int J Cancer. 2012;131:E337-47
24. Israelsen WJ, Vander Heiden MG. ATP consumption promotes cancer metabolism. Cell. 2010;143:669-71
25. Wang Q, Liang B, Shirwany NA, Zou MH. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One. 2011;6:17234
26. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441-64
27. Maher JC, Savaraj N, Priebe W, Liu H, Lampidis TJ. Differential sensitivity to 2-deoxy-D-glucose between two pancreatic cell lines correlates with GLUT-1 expression. Pancreas. 2005;30:34-9
28. Coleman MC, Asbury CR, Daniels D, Du J, Aykin-Burns N, Smith BJ. et al. 2-deoxy-D-glucose causes cytotoxicity, oxidative stress, and radiosensitization in pancreatic cancer. Free Radic Biol Med. 2008;44:322-31
29. Bu HQ, Luo J, Chen H, Zhang JH, Li HH, Guo HC. et al. Oridonin enhances antitumor activity of gemcitabine in pancreatic cancer through MAPK-p38 signaling pathway. Int J Oncol. 2012;41:949-58
30. Ding XZ, Adrian TE. MEK/ERK-mediated proliferation is negatively regulated by P38 map kinase in the human pancreatic cancer cell line, PANC-1. Biochem Biophys Res Commun. 2001;282:447-53
31. Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K. et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669-76
32. Landau BR, Laszlo J, Stengle J, Burk D. Certain metabolic and pharmacologic effects in cancer patients given infusions of 2-deoxy-D-glucose. J Natl Cancer Inst. 1958;21:485-94
33. Blouin MJ, Zhao Y, Zakikhani M, Algire C, Piura E, Pollak M. Loss of function of PTEN alters the relationship between glucose concentration and cell proliferation, increases glycolysis, and sensitizes cells to 2-deoxyglucose. Cancer Lett. 2010;289:246-53
34. Rodriguez-Enriquez S, Gallardo-Perez JC, Marin-Hernandez A, Moreno-Sanchez R. The Warburg Hypothesis and the ATP Supply In Cancer Cells Is Oxidative Phosphorylation impaired in malignant neoplasias? Curr Pharm Biotechnol. 2012
35. Freinkel N, Metzger BE, Harris E, Robinson S, Mager M. The hypothermia of hypoglycemia. Studies with 2-deoxy-D-glucose in normal human subjects and mice. N Engl J Med. 1972;287:841-5
36. Wu H, Zhu H, Liu DX, Niu TK, Ren X, Patel R. et al. Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res. 2009;69:2453-60
37. Com E, Lagadec C, Page A, El Yazidi-Belkoura I, Slomianny C, Spencer A. et al. Nerve growth factor receptor TrkA signaling in breast cancer cells involves Ku70 to prevent apoptosis. Mol Cell Proteomics. 2007;6:1842-54
38. Gezginci-Oktayoglu S, Bolkent S. 4-Methlycatechol prevents NGF/p75(NTR)-mediated apoptosis via NGF/TrkA system in pancreatic beta cells. Neuropeptides. 2011;45:143-50
39. Lagadec C, Meignan S, Adriaenssens E, Foveau B, Vanhecke E, Romon R. et al. TrkA overexpression enhances growth and metastasis of breast cancer cells. Oncogene. 2009;28:1960-70
Author contact
![]() Corresponding author: Cheng-Xue Dang, PhD, the Department of surgical oncology the First Affiliated Hospital, Xi'an Jiaotong University College of Medicine, 277 W. Yanta Road, Xi'an, Shaanxi 710061, China. E-mail: dangchengxuecom.cn
Corresponding author: Cheng-Xue Dang, PhD, the Department of surgical oncology the First Affiliated Hospital, Xi'an Jiaotong University College of Medicine, 277 W. Yanta Road, Xi'an, Shaanxi 710061, China. E-mail: dangchengxuecom.cn