Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(5):522-531. doi:10.7150/ijms.5325 This issue Cite
Research Paper
IGF-1 Acts as Controlling Switch for Long-term Proliferation and Maintenance of EGF/FGF-responsive Striatal Neural Stem Cells
Nor Entan Supeno1*, Soumya Pati1*, Raisah Abdul Hadi1, Abdul Rahman Izani Ghani1, Zulkifli Mustafa1, Jafri M Abdullah1, Fauziah Mohamad Idris2, Xu Han3, Hasnan Jaafar4 ![]()
1. Department of Neurosciences, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia;
2. Department of Medical Microbiology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia;
3. Department of Pediatrics, Baylor College of Medicine, TX 77030, USA;
4. Department of Pathology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia.
* Co-first authors.
Received 2012-10-1; Accepted 2013-3-5; Published 2013-3-13
Abstract
Background: Long-term maintenance of neural stem cells in vitro is crucial for their stage specific roles in neurogenesis. To have an in-depth understanding of optimal conditional microenvironmental niche for long-term maintenance of neural stem cells (NSCs), we imposed different combinatorial treatment of growth factors to EGF/FGF-responsive cells. We hypothesized, that IGF-1-treatment can provide an optimal niche for long-term maintenance and proliferation of EGF/FGF-responsive NSCs.
Objective: This study was performed to investigate the cellular morphology and growth of rat embryonic striatal tissue derived-NSCs in long-term culture under the influence of different combinatorial effects of certain growth factors, such as EGF, bFGF, LIF and IGF-1.
Methods: The NSCs were harvested and cultured from striatal tissue of 18 days old rat embryos. We have generated neurospheres from these NSCs and cultured them till passage 7 (28 days in vitro) under four different conditional microenvironments: (A) without growth factor, (B) EGF/bFGF, (C) EGF/bFGF/LIF, (D) EGF/bFGF/IGF-1 and (E) EGF/bFGF/LIF/IGF-1. Isolated NSCs were characterised by Immunoflouroscence for nestin expression. The cell growth and proliferation was evaluated at different time intervals (P1, P3, P5 & P7), assessing the metabolic activity based cell proliferation. Apoptosis was studied in each of these groups by In situ cell death assay.
Results: Our results demonstrated certain important findings relevant to long-term culture and maintenance of striatal NSC-derived neurospheres. This suggested that IGF-1 can induce enhanced cell proliferation during early stages of neurogenesis, impose long-term maintenance (up to passage 7) to cultured NSCs and enhance survival efficiency in vitro, in the presence of EGF and FGF.
Conclusions: Our findings support the hypothesis that the enforcement of IGF-1 treatment to the EGF/FGF-responsive NSCs, can lead to enhanced cell proliferation during early stages of neurogenesis, and an extended life span in vitro. This information will be beneficial for improving future therapeutic implication of NSCs, by addressing improved in vitro production of NSCs.
Keywords: neural stem cells, neurospheres, proliferative activity, EGF, bFGF, LIF, IGF-1.
Introduction
Current stem cell based regenerative therapies include a broad range of adult stem cell sources like; neural stem cells (NSCs) and mesenchymal stem cells (MSCs) in neural repair [30]. In recent years, there has been growing interest in the use of neural stem cells (NSCs) as a novel approach for the treatment of neurodegenerative diseases. NSCs have gained popularity as a potential cell therapy agent due to their ability to mitigate neurodegenerative diseases by replacing impaired tissues.
NSCs are characterised as undifferentiated cells with the capacity to self-renew (while maintaining an undifferentiated state) that can give rise to all neural lineages: including neurons, astrocytes and oligodendrocytes [3, 8, 32, 36, 41]. The major pitfall that has been encountered till now in growing NSCs is their long-term maintenance in vitro, due to unavailability of optimal guidance cues for their microenvironmental niche.
Studies in recent past have demonstrated certain neurotrophins like; Insulin growth factor (IGF-1), Fibroblast Growth Factor (FGF), Epidermal Growth Factor (EGF) and Leukaemia Inhibitory Factor (LIF) can induce recovery of post-ischemic brain injury in rats [13, 19, 24, 28, 34]. Among these factors, recruitment of the dual colonial growth factors; such as FGF-2 and EGF was found to be essential for growth and survival of E14-E20 embryonic striatal stem cells (ESSCs) [14]. Previous studies have also shown that Insulin Growth Factor-1 (IGF-1) can affect early embryonic development in culture by increasing the total cell number [11, 18]. Interestingly, a group of evidences also suggested that IGF-1 plays a controlling factor for the above factors strictly regulating their phenotypic abilities [2]. And LIF could increase proliferation and telomerase expression of EGF-responsive human fetal cortical neural progenitors in vitro, but with decreased neurogenesis [38].
Based on these experimental evidences, we designed an experimental strategy to investigate the effect of IGF-1 and LIF on long-term maintenance of EGF/FGF- responsive embryonic striatal neural precursors from E18 rat embryos, as both EGF/FGF are essential for in vitro culture of NSCs. Our findings have suggested that administration of IGF-1 to EGF/FGF-responsive striatal progenitors can enhance proliferating ability of these cells in long-term culture in vitro. This combinatorial microenvironment will be extremely beneficial for future NSC-based therapies in neuro-regenerative application.
Material and methods
Breeding of Sprague-Dawley (SD) rat
Monogamous pairs of male (8-10 weeks old) and female SD rats (10-14 weeks old) were obtained from the Animal House Unit (LARAS, USM). A healthy SD male rat was housed individually in a new cage for one to two hours to ensure proper adaptation, prior to introduction of a healthy female SD rat into the same cage. The animals were left overnight, following a stable social interaction (non-fighting mode), and the female rat was checked for pregnancy the next morning using the vaginal swab method [12]. The caging and handling of the rats were carried out in accordance with proper laboratory practice criteria as defined by the Animal House Unit (LARAS, USM), and all experimentation was approved by the Animal Ethics Committee (Number of approval: USM/Animal Ethics Approval/2007/30/097).
Isolation of embryonic rat striatal tissue
The isolation of embryonic striatal tissue was carried out as previously described [1, 10, 15, 22]. Pregnant SD rats with embryos aged E18 days of gestation were selected for the experiment. The pregnant rat was anesthetised using a mixture of ketamine/xylazine at dosages ranging from 40 to 87 mg/kg for ketamine and 5 to 13 mg/kg for xylazine. The peritoneal area was cleaned with hibiscrub, shaved, wiped with 70% alcohol and finally wiped with iodine for aseptic purposes. The rat was then covered with fenestrated sterile gauze, except for three inches of the peritoneal midline, which was left exposed. A midline incision was performed on the skin as the uterus and the embryos were aseptically removed. The embryos were harvested from their individual sacs beginning at the distal end of the uterine horn. The pregnancy stage of the rats was estimated by measuring the crown-rump length (CRL) of the embryos as previously described [10]. Each embryo was immersed in 70% ethanol for one second to reduce bleeding and to avoid sources of contamination from the blood. The embryo was then placed into a petri dish containing cold 6% glucose in PBS (PBSg) to keep the brain tissue moist. The embryonic striatal tissue was isolated as previously described [8]. Using a stereoscopic dissecting microscope, a striatal surface region was anatomically identified by its characteristic blotchy appearance. The striatal tissues were then isolated and pooled into a 50 mL falcon tube containing 15 mL of PBSg, which was followed by the culturing procedures. The pooled striatal tissues were minced into small sections and 1% penicillin/streptomycin were added to prevent contamination.
Culturing of NSCs
The methods used to culture the embryonic striatal tissue were performed as previously described [5] with some modifications. Briefly, the pooled striatal tissue in phosphate buffer saline with glucose, PBSg was rinsed in the presence of 1% penicillin/streptomycin. The tissues were digested by adding 1% detachin for 10 to 20 minutes in a 37ºC water bath. The cells were then centrifuged at 1600 rpm for 5 minutes followed by the pellets were re-suspended in 1 mL of serum-free media. Single-cell suspensions were obtained using mechanical dissociation. The mixture was triturated using a 23G syringe needle and filtered through a 40 µm cell strainer to remove debris and non-dissociated cells. Cells were counted using trypan blue to verify cell viability. The cells were then grown using the Neurocult NS-A Proliferation Media kit for rat (StemCell Technologies, Inc, USA, C/N: 05771). In accordance with the study design, EGF (20 ng/mL), bFGF (20 ng/mL), LIF (10 ng/mL), IGF-1 (100 ng/mL) and/or heparin (5 µg/mL) were added to the media. In this study, the NSCs were cultured in five experimental groups: group A (without growth factor), group B (EGF/bFGF), group C (EGF/bFGF/LIF), group D (EGF/bFGF/IGF-1) and group E (EGF/bFGF/LIF/IGF-1). The cells were cultured in T-25 cm2 culture flasks at a cell density of 10-15 cells/µL, and the growth factors were added every two to three days. Neurospheres usually formed after three to four days of in vitro culturing. The culture flasks were gently agitated several times a day to prevent adherence of the neurospheres to the flask wall, which would result in cell differentiation. The culture flasks were maintained and incubated at temperature of 37ºC using 5% CO2 incubator.
Passaging of NSCs
Following confluency, the neurospheres were passaged into different flasks in order to promote expansion of cell number. The neurospheres were passaged every 4th day in vitro, once they reach the size of 150 to 200 µm in diameter, as they can be easily dissociated into single cells at this stage. These neurospheres were then centrifuged at 400 rpm for 5 minutes, followed by 1600 rpm for 6 minutes to separate the cells from media [40]. The supernatant was gently removed, and the cell pellets were then incubated in 1% detachin for 10 minutes at 37ºC. After incubation, the neurospheres were triturated first using a pasteur pipette, then using a 1 mL pipette tip and finally with a 23G syringe needle, 10 to 15 times for each needle, as previously described [17]. These cells were again centrifuged at 1600 rpm for 5 minutes, to discard the supernatants. Finally, the pellet containing single cells were re-seeded in complete medium containing growth factors.
Cellular morphology of the NSCs
The cellular morphology of the NSCs was examined during four different passages: P1, P3, P5 and P7 (each passage was performed at every four days in vitro). We compared the cellular morphology of the NSCs and examined the sizes of the neurospheres from the five different growth factor groups. In this way, we determined the optimal combination of growth factors with respect to NSC cell growth and cellular morphology. Cellular morphology was assessed by observation under an inverted microscope.
Immunocytochemical staining
Immunocytochemical (ICC) staining was performed to ensure that the isolated cells were indeed neural stem cells. In this study, adult rat hippocampal neural stem cells were used as a positive control (Chemicon International, Inc, USA, C/N: SCR022). Characterisation of the NSCs was performed as follows: Neurospheres were placed into 8-well chamber slides. The cells were then fixed using 4% paraformaldehyde in 1X PBS and incubated for 30 to 40 minutes at RT. After incubation, the fixative was carefully removed using aspiration, and the cells were washed three times (5 to 10 minutes each) with 1X PBS. After washing, the cells were permeabilised with 0.3% Triton X-100. Next, cells were washed with a blocking solution of 5% normal donkey serum (Chemicon International, Inc, USA, C/N: S30-100ML) for at least two hours at RT or overnight at 4ºC. The cells were then incubated overnight at 4ºC with the primary antibody mouse anti-nestin (monoclonal, 1:200) (Chemicon International, Inc, USA, C/N: 2003602) [24, 30]. Negative controls were treated identically except for the addition of the primary antibody. The next day, cells were washed twice with 1X PBS and twice with blocking solution. After the last wash step, the cells were left in blocking solution for at least 30 minutes. Donkey anti-mouse IgG conjugated to Cy3 (1:250 or 1:500) (Chemicon International, Inc, USA, C/N: AP192C) was used as the secondary antibody, and the cells were incubated with this secondary antibody for two hours at RT. After incubation, the cells were washed for three to five times using 1X PBS. Cell nuclei were then stained with acridine orange at a working concentration of 1:500. Finally, the sections were mounted on glass chamber slides using anti-fading UltraCruzTM mounting medium to minimise photo-bleaching after prolonged exposure to a high-intensity light source. The fixed cells were then visualised using a Pascal 5 confocal microscope (Carl Zeiss, Germany).
Proliferation study
Proliferation studies were performed using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega Corporation, USA, C/N: G3580), which uses a novel tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS (a)] and a chemically stable electron coupling reagent (phenazine ethosulfate; PES) to form a stable “One Solution”. Bioreduction of MTS to colored formazan was read at 490nm, to evaluate metabolically active cells, and the level of cytotoxicity in cutured cells. Our experimental strategy involved five groups of combinatorial growth factor conditions (group A:without growth factor, group B: EGF/bFGF, group C: EGF/bFGF/LIF, group D: EGF/bFGF/IGF-1 and group E: EGF/bFGF/LIF/IGF-1). NSCs were grown at a starting density of 1X103 cells per well of 96-well culture plate, with 5 replicates for each group. Spectrophotometric recordings were obtained from each group of cells at 0, 24, 48, 72 and 96 hours following the exact procedure suggested by instruction manual, to evaluate the increment in cell proliferation.
In situ cell death detection (TUNEL)
To demonstrate the effect of different neurotrophins on NSCs undergoing apoptosis, we used a terminal dUTP nick-end labelling (TUNEL) assay. At 12 days in vitro, neurospheres were triturated into single cells and plated on poly-L-lysine-coated 8-well chamber slides (BD BioCoat, USA) at a concentration of 50,000 per ml per well and incubated for an additional 6 h at 37°C in a 5% CO2 incubator. The apoptotic cells were labelled according to the instructions of the in situ Cell Death Detection Kit (Roche, Penzberg, Germany). The TUNEL-labelled cells were further counter-stained and mounted using Ultra Cruz mounting dye containing DAPI (Santa Cruz). The number of TUNEL+ NSCs was detected by Zeiss fluorescence microscopy and normalised to DAPI-stained cells.
Statistical analysis
Two-way anova was performed among cultured NSCs at P1, P3, P5 and P7 for different time intervals; such as 24hrs, 48hrs, 72hrs and 96hrs, to elucidate the changes in cell proliferation. The NSCs from each group were also compared using pair wise comparisons using Tukey's HSD test (Tukey's honestly significant difference test). Based on this analysis, we concluded that time and cell types derived from each group, led to a significant change in values. Consequently, we also considered these main factors separately to explore how the values changed in response to each of these two factors. To achieve this goal, we separated our comparisons at each time point. Because time interval was fixed, we applied Tukey's HSD test (Tukey's honestly significant difference test) to compare the means of NSCs from two different conditions. The result was represented by a colored matrix. Each box in this matrix represented a paired comparison, and the matrix is colour coded by P-values ranging from 0 to 1 showing red as low and blue as high. The P-values for the paired comparisons were recorded for each group, to clearly delineate the significantly different pairs in the matrix. All data are expressed as the mean ± SD. P-values <0.05 were considered statistically significant in all experiments.
Results
Cellular morphological analysis
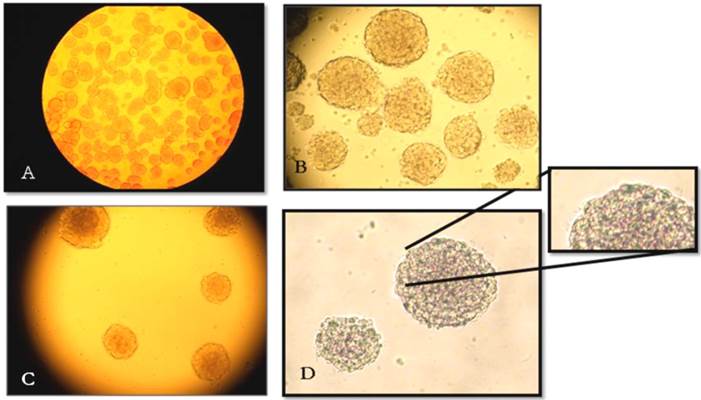
The cellular morphology of striatum-derived progenitors were analysed by microscopy for changes in appearance during each passage (P1, P3, P5 and P7). We observed that new floating spheres were formed as a consequence of cell proliferation. These cellular aggregates or neurospheres were identified as both well-defined spherical cell clusters as well as more irregular cell clusters, as shown in Figure 1. Cilia were also present on the surface or outer edges of some of the neurospheres, indicating that the neurospheres were in a healthy condition, as previously reported [5, 18].
Cellular morphological analysis of neurospheres. (A) Neurospheres vary in shape; some are well-formed spheres, whereas others are irregular cell clusters. (B) The P1 of NSCs cultures contained cell debris. (C) By P3, neurosphere cultures contained less debris compared with P1. The neurospheres adopted spherical shapes with bright surfaces. (D) Healthy neurospheres are composed of many individual cells and possess small cilia that are apparent on the outer edge. All images were viewed at 100X magnification using an inverted microscope.
Immunocytochemical staining of NSCs
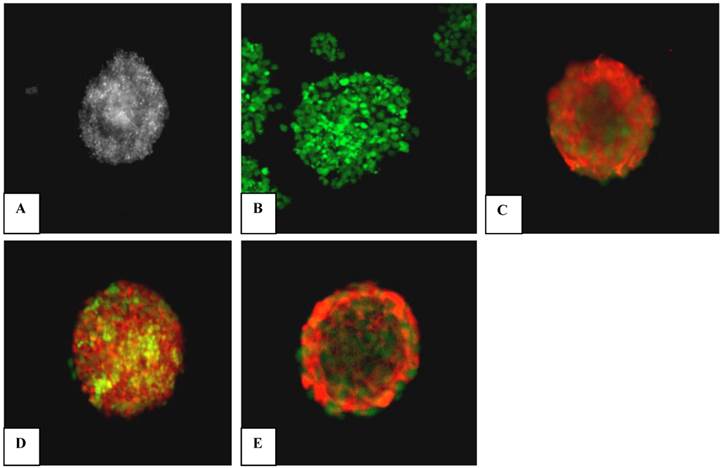
NSCs are represented by expression of Nestin; a most prominent signature associated with neural precursors of central nervous system [32]. Immunocytochemical staining of NSCs was performed to characterise the isolated cell population. Our findings show that neurosphere isolates from all the experimental groups were positively stained for nestin at P1, as shown in Figure 2. The isolated neurospheres reached confluency between 3 to 5 days in vitro. Nestin characterization was performed for neurospheres at P1 and P3 stages. The results displayed that neurospheres at P3 (Figure 2) stained positive for nestin and appeared as well-defined spheres compared to P1. Our results supported a previous report, which suggested that NSCs during early passage (e.g., P1) contain numerous dead cells and cell debris that can be eliminated by further passaging [16].
Immunocytochemical staining of embryonic striatal stem cells in culture. Results show that neurospheres were positively stained for nestin (red). (A) Phase-contrast image of a neurosphere. (B) Negative control. (C) Adult rat hippocampal neural stem cells as positive control. (D) Neurosphere at P1. (E) Neurosphere at P3. Neurospheres were viewed at 200X magnification using a Pascal 5 confocal microscope (Carl Zeiss, Germany).
Long term maintenance of neurospheres in vitro
In this study, we have evaluated the relative sizes of NSCs from the four different combinatorial groups of growth factors: e.g., group A (without growth factor), group B (EGF/bFGF), group C (EGF/bFGF/LIF), group D (EGF/bFGF/IGF-1) and group E (EGF/bFGF/LIF/IGF-1) at four passages (P1, P3, P5 and P7). These neurospheres were able to remain as undifferentiated spheres till P7. Additionally, the amount of debris was reduced following continuous passaging in all of the groups. We also found that the NSC spheres grew to different sizes in the different growth factor groups. For example, our results show that neurospheres from group D were relatively larger and mature than those from the other groups during all of the examined passages (Figure 3). On the other hand, group A had the smallest spheres during all passages. Interestingly, group D neurospheres from P3 to P7 showed the greatest confluency only three days after subculture, in contrast with the other groups, but with a much denser core as compared to other groups.
The NSCs at P1 until P7 shows different sizes of neurospheres grown in different groups of growth factors. Group A (without growth factor); Group B (EGF/bFGF); Group C (EGF/bFGF/LIF); Group D (EGF/bFGF/IGF-1) and group E (EGF/bFGF/LIF/IGF-1). Group D had the largest in size of neurospheres. Viewed at 100X magnification using an inverted microscope.

Effect of IGF-1 on proliferation and survival of NSCs
To understand the role of long term proliferation and survival efficiency of neurospheres in vitro, we have grown EGF/FGF responsive NSCs alone and under the influence of different combinations of growth factors until passage 7. To elucidate the effects of these factors on proliferation of NSCs, we have investigated the changes in their metabolic viability at different time intervals (24 hrs, 48 hrs, 72 hrs and 96 hrs) till P7, using a spectrophotometric based cell proliferation assay in vitro. This assay demonstrated a visual representation of changes in the number of metabolically viable cells among different treatment groups, by evaluating the metabolic activity of cellular enzymes that converted the tetrazolium dye MTS ([3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) into formazan product. So the amount of formazan production not only indicated both growth and proliferation of NSCs, also showed the effect of growth factor treatments on NSC cytotoxicity.
Our results suggested three important findings; 1) IGF-1 enhanced proliferation of neural stem cells during the early stages of neurogenesis in vitro (Fig. 4A); 2) IGF-1-I treated EGF/FGF responsive population showed higher survival efficiency as shown by less tunnel positive cells (Fig. 4B); 3) LIF treatment couldn't rescue the EGF/FGF responsive cells even in the presence of IGF-1 (Fig. 4A).
Long-term Proliferation and survival of ESSCs: Embryonic neural stem cells were cultured under different cocktails of growth factors (n=5) and change in proliferation was studied at P1, P3, P5 and P7 days of passage using in vitro cell proliferation assay (A).Two-way anova analysis was performed for all the above cell populations to study the significance changes in the rate of proliferation. The data was represented as optical density (OD) at 540nm in mean ± SD. P-value <0.05 was considered as significant. Survival efficiency was also evaluated for ESSCs from all groups by TUNEL assay, and staining was visualized in fluorescent microscopy at 20X magnification. Panel B demonstrated TUNEL/DAPI positive NSCs at P3 from different niche conditions (B). Data only shown for group C and group D derived NSCs. The TUNEL/DAPI positive NSCs from control group A cultured without any growth factor.
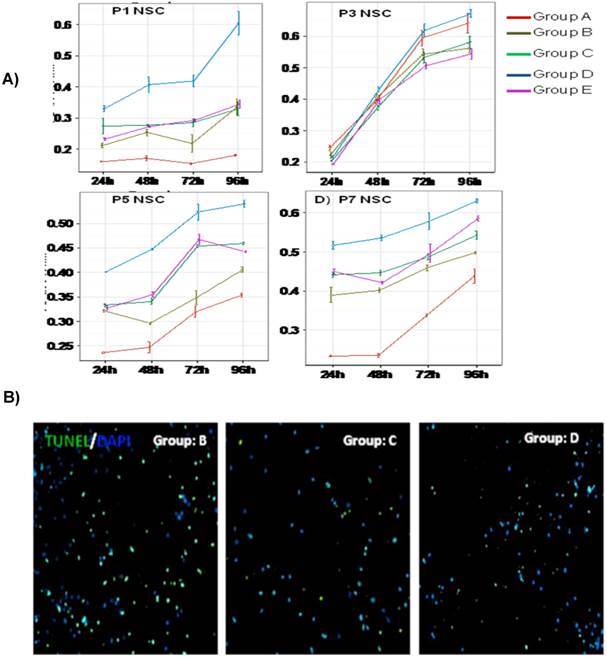
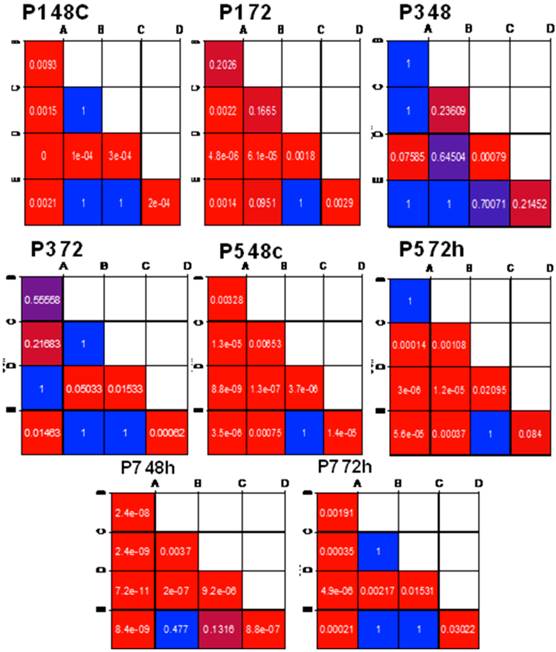
To validate these data we have also performed a two-way ANOVA analysis among different groups of NSCs, which demonstrated that IGF-1 treatment could lead to a stable increase in cell proliferation at almost all time points (24 hrs, 48 hrs, 72 hrs and 96 hrs) (n=5,* P ≤0.05) during the early stages of development (Fig.4A). To further compare the substantial changes in proliferation of IGF-instructed NSCs with other groups at different time intervals (P1, P3, P5 and P7), we have also performed Pair wise comparative analysis (Fig.5). These results also explained the possible significance of growth factor treatments on long-term growth profile of NSCs in vitro.
Pair wise comparative analysis of long-term growth profile of ESSCs at different time intervals: Pair wise comparison was analyzed in detail, among NSCs cultured under the influence of different growth factor conditions at P1, P3, P5 and P7 passages using Tukey's HSD (Tukey's honestly significant difference test). The data was represented as mean ± SD and statistical significance was based on P-value <0.05.
Our data also demonstrated an interesting observation, which suggested that addition of LIF to the culture was detrimental to the early stage of proliferation of NSCs, as shown by reduced proliferation (Fig.4A). And even the treatment of IGF-1 could not rescue the proliferation of NSCs (Fig.4A).
Discussion
Accumulating evidence from adult neurogenesis-based studies has shown that synergistic treatment with FGF-2 and EGF increases progenitor proliferation in the sub-ventricular zone (SVZ) of global ischemia and enhances post-ischemic memory formation and retention [4, 30]. Both EGF and FGF-2 are co-localised during embryonic developmental stages and comprise a synergistic signalling pathway in neural stem cells, which significantly contributes to neurogenesis. It was also noteworthy those IGF-1 acts as a controlling switch for both of these “colonial” growth factors, and modulates their action during neurogenesis [2].
Our study has strongly corroborated the above information, as IGF-1 treatment could drastically enhance early stage proliferation of EGF/FGF-responsive neural stem cells in vitro (Fig. 4A). Although the enhancement in proliferation was maintained till P7, the change was most evident in the early stages at P1 and P3 (Fig. 4A). Earlier findings have shown that IGF-1 acts via the extracellular signal-related kinase (ERK) /mitogen-activated protein kinase (MAPK) pathway, activating cAMP response element-binding protein (CREB) and leading to enhanced mitogenesis [22]. Our data has indicated a positive regulatory role of IGF-1, as demonstrated by the consistency in enhancement proliferation of IGF-1-treated NSCs during the early stages of neurogenesis. Our results also presented another interesting piece of information, which suggested that, treatment of LIF can negatively attenuate early stage proliferation of EGF/FGF-responsive NSCs, and could not increase the survival efficiency of these cells, even in presence of IGF-1 (Fig. 4A).
These results led us to hypothesize that 1) LIF could not support the proliferation of NSCs, and 2) it might regulate the early-stage maturation of these cells. Our hypothesis directly corroborates some earlier published reports, which suggested, LIF treatment could not enhance the survival of cortical precursor cells isolated from later rat embryos (beyond embryonic day 16, E16) and might also regulate the number of neural precursors in the stem cell pool by preventing the emergence of more differentiated cell types via the activation of the transcription factor STAT3 [12,27]. Thus our results explained, LIF treatment in NSCs could not help recovery of NSCs isolated from E18 embryos and also might attenuate the maturation of NSCs to maintain the self-renewing pool of NSCs. Based on these observations it is important to highlight that, an optimal microenvironmental niche containing growth factors like IGF-1+EGF+FGF can be important in long-term maintenance and growth of embryonic neural stem cells in vitro.
Conclusion
We hereby conclude that IGF-1 can be the important guidance cue for EGF/FGF-responsive neural stem cells in vitro, and play crucial role in long-term proliferation and maintenance of neural stem cells.
Acknowledgements
This study was supported by the Fundamental Research Grant Scheme (FRGS), Ministry of Higher Education, Malaysia (Grant No. 203/PPSP/6171106).
Competing interests
The authors declare that no competing interests exist.
References
1. Altman J, Bayer SA. Atlas of prenatal rat brain development. USA: CRC Press Ltd, London. 1995
2. Arsenijevic Y, Weiss S, Schneider B, Aebischer P. et al. Insulin-Like Growth Factor-I Is Necessary for Neural Stem Cell Proliferation and Demonstrates Distinct Actions of Epidermal Growth Factor and Fibroblast Growth Factor-2. J Neurosci. 2001;21(18):7194-7202
3. Bazan E, Alonso FJM, Redondo C, Lopez MAT, Alfaro JM, Reimers D, Herranz AS, Paino CL, Serrano AB, Cobacho N, Caso E, Lobo MVT. et al. In vitro and in vivo characterization of neural stem cells. Histology and Histopathology: Cell Mol Biol. 2004;19(4):1261-1275
4. Blomeley CP, Cains S, Smith R, Bracci E. et al. Ethanol Affects Striatal Interneurons Directly and Projection Neurons Through a Reduction in Cholinergic Tone. Neuropsychopharmacology. 2011;36(5):1-14
5. Bez A, Corsini E, Curti D, Biggiogera M, Colombo A, Nicosia RF, Pagano SF, Parati EA. et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 2003;993(1-2):18-29
6. Cai J, Cheng A, Luo Y, Lu C, Mattson MP, Rao MS, Furukawa K. et al. Membrane properties of rat embryonic multipotent neural stem cells. J Neurochem. 2004;88(1):212-226
7. Castillo L, Martinez L, Grygar E, Hutter-Paier B. et al. Characterization of proliferation and differentiation of EGF-responsive striatal and septal precursor cells. Int J Dev Neurosci. 2003;21(1):41-47
8. Choi KC, Yoo DS, Cho KS, Huh PW, Kim DS, Park CK. et al. Effect of single growth factor and growth factor combinations on differentiation of neural stem cells. J Korean Neurosurg Soc. 2008;44(6):375-381
9. Covey MV, Levison SW. Leukemia inhibitory factor participates in the expansion of neural stem/progenitors after perinatal hypoxia/ischemia. Neuroscience. 2007;148(2):501-509
10. Dunnett SB, Björklund A. Dissecting embryonic neural tissues for transplantation. In: (ed.) Dunnett SB, Boulton AA, Baker GB. Neural transplantation methods. USA: Humana Press Inc. 2000:3-25
11. Fabian F, Il'kova G, Reha KP, Czikkova S, Baran V, Koppel J. et al. Inhibitory effect of IGF-1-I on induced apoptosis in mouse preimplantation embryos cultured in vitro. Theriogenology. 2004;61(4):745-755
12. Jeffrey JL, Sonya PS. Reproduction and breeding. Mark AS, Steven HW, Craig LF. 2nd ed. The laboratory rat. USA: Elsevier Academic Press publication. 2006:147-164
13. Jin K, LaFevre-Bernt M, Sun Y, Chen S, Gafni J, Crippen D, Logvinova A, Ross CA, Greenberg DA, Ellerby LM. et al. FGF-2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington's disease. Proc.Natl.Acad.Sci. 2005;102(50):18189-18194
14. Kelly CM, Tyers P, Borg M, Svendsen CN, Dunnett SB, Rosser AE. et al. EGF and FGF-2 responsiveness of rat and mouse neural precursors derived from the embryonic CNS. Brain Resh Bull. 2005;68(1-2):83-94
15. König N, Wilkie MB, Lauder J. et al. Dissection of monoaminergic neuronal groups from embryonic rat brain. In: (ed.) Shahar A, Vellis J, Vernadakis A, Haber B. 2nd ed. A dissection and tissue culture manual of the nervous system. USA: Alan R. Liss, Inc. 1990:26-29
16. Laura PKK, Shelley S, Jacqueline GA, Kasia T, Laurie DC. Neural Stem Cell Culture: Neurosphere generation, microscopical analysis and cryopreservation. Protocol Exchange 2006; doi:10.1038/nprot. 2006 215. http://www.nature.com/protocolexchange/protocols/77
17. Li X, Xu J, Bai Y, Wang X, Dai X, Liu Y, Zhang J, Zou J, Shen L, Li L. et al. Isolation and characterization of neural stem cells from human fetal striatum. Biochem Biophys Res Com. 2005;326(2):425-434
18. Lin TC, Yen JM, Gong KB, Hsu TT, Chen LR. et al. IGF-1/IGF-1BP-1 increases blastocyst formation and total blastocyst cell number in mouse embryo culture and facilitates the establishment of a stem-cell line. BMC Cell Biol. 2003;4(14):1-15
19. Lin S, Fan LW, Rhodes PG, Cai Z. Intranasal administration of IGF-1 attenuates hypoxic- ischemic brain injury in neonatal rats. Exp Neurol. 2009;217(2):361-70
20. Lobo MVT, Alonso FJM, Redondo C, Lopez-Toledano MA, Caso E, Herranz AS, Paino CL, Reimers D, Bazan E. et al. Cellular characterization of epidermal growth factor-expanded free-floating neurospheres. J Histochem Cytochem. 2003;51(1):89-103
21. Louis SA, Reynolds BA. Neurosphere and neural colony-forming cell assays. Doering, L. C. 4th edition. Protocols for neural cell culture. USA: Humana Press Inc. 2010:1-28
22. Messer A. Dissection of newborn rat corpus striatum. Shahar A, Vellis J, Vernadakis A, Haber B, et al. 2nd ed. A dissection and tissue culture manual of the nervous system. United States of America: Alan R. Liss, Inc. 1990:23-25
23. Moon C, Yoo JY, Matarazzo V, Sun YK, Kim EJ, Ronnett GV. et al. Leukemia inhibitory factor inhibits neuronal terminal differentiation through STAT3 activation. Proc.Natl.Acad.Sci. 2002;99(13):9015-9020
24. Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 2002;110(4):429-441
25. Pardo B, Honegger P. Differentiation of rat striatal embryonic stem cells in vitro: Monolayer culture vs. three-dimensional coculture with differentiated brain cells. J Neurosci Res. 2000;59(4):504-512
26. Raballo R, Rhee J, Lyn RC, Leckman JF, Schwartz ML, Vaccarino FM. et al. Basic Fibroblast Growth Factor (FGF-2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci. 2000;20(13):5012-5023
27. Roobrouck VD, Ulloa FM, Verfaillie CM. et al. Self-renewal and differentiation capacity of young and aged stem cells. Exp Cell Res. 2008;314(9):1937-1944
28. S. Bauer and P. H. Patterson: The cell cycle-apoptosis connection revisited in the adult brain. J Cell Biol. 2005;171(4):641-650
29. Singec I, Knoth R, Meyer RP, Maciaczyk J, Volk B, Nikkhah G, Frotscher M, Snyder EY. et al. Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat Meth. 2006;3(10):801-806
30. Shimada IS, Spees JL. Stem and progenitor cells for neurological repair: Minor issues, major hurdles, and exciting opportunities for paracrine-based therapeutics. J Cell Biochem. 2011;112:474-380
31. Smith R, Bagga V, Fricker-Gates RA. et al. Embryonic neural progenitor cells: The effects of species, region, and culture conditions on long-term proliferation and neuronal differentiation. J. Hematother. Stem Cell Res. 2003;12(6):713-725
32. Sun T, Wang XJ, Xie SS, Zhang DL, Wang XP, Li BQ, Ma W, Xin H. et al. A comparison of proliferative capacity and passaging potential between neural stem and progenitor cells in adherent and neurosphere cultures. J Dev Neurosci. 2011;29(7):723-731
33. Tarasenko YI, Yu Y, Jordan PM, Bottenstein J, Wu P. et al. Effect of growth factors on proliferation and phenotypic differentiation of human fetal neural stem cells. J Neurosci Res. 2004;78(5):625-636
34. Tureyen K, Vemuganti R, Bowen KK, Sailor KA, Dempsey RJ. EGF and FGF-2 infusion increases post-ischemic neural progenitor cell proliferation in the adult rat brain. Neurosurgery. 2005;57(6):1254-1263
35. Vukicevic V, Jauch A, Dinger TC, Gebauer L, Hornich V, Bornstein SR, Ehrhart MB, Muller AM. et al. Genetic instability and diminished differentiation capacity in long-term cultured mouse neurosphere cells. Mechanisms of Ageing and Development. 2010;131(2):124-132
36. Wang TY, Sen A, Behie LA, Kallos MS. et al. Dynamic behavior of cells within neurospheres in expanding populations of neural precursors. Brain Res. 2006;1107(1):82-96
37. Wright LS, Prowse KR, Wallace K, Linskens MHK, Svendsen CN. et al. Human progenitor cells isolated from the developing cortex undergo decreased neurogenesis and eventual senescence following expansion in vitro. Exp Cell Res. 2006;312(11):2107-2120
38. Wright LS, Li J, Caldwell MA, Wallace K, Johnson JA, Svendsen CN. et al. Gene expression in human neural stem cells: effects of leukemia inhibitory factor. J Neurochem. 2003;86(1):179-195
39. Xue T, Qiao Li, Lu L, Zha D, Wei L. et al. Proliferation, multipotency and neuronal differentiation of cryopreserved neural progenitor cells derived from the olfactory neuroepithelium of the adult rat. Cell Biol Int. 2008;32(8):950-958
40. Ye S, Su Z, Zhang J, Qian X, Zhuge Q, Zeng Y. et al. Differential centrifugation in culture and differentiation of rat neural stem cells. Cell Mol Neurobiol. 2008;28(4):511-517
41. Yun SJ, Byun K, Bhin J, Oh JH, Nhung LTH, Hwang D, Lee B. et al. Transcriptional regulatory networks associated with self-renewal and differentiation of neural stem cells. J Cell Physiol. 2010;225(2):337-347
Author contact
![]() Corresponding author: Dr. Hasnan Jaafar. E-Mail: hasnanusm.my Telephone: +609-7676300 or +609-7676466 Fax: +609-7653370.
Corresponding author: Dr. Hasnan Jaafar. E-Mail: hasnanusm.my Telephone: +609-7676300 or +609-7676466 Fax: +609-7653370.