ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2013; 10(4):420-426. doi:10.7150/ijms.5723 This issue Cite
Research Paper
Inflammatory Stress Exacerbates the Progression of Cardiac Fibrosis in High-Fat-Fed Apolipoprotein E Knockout Mice via Endothelial-Mesenchymal Transition
Kun Ling Ma1, Jing Liu1, Jie Ni1, Yang Zhang1, Lin Li Lv1, Ri Ning Tang1, Hai Feng Ni1, Xiong Zhong Ruan2, Bi Cheng Liu1 ![]()
1. Institute of Nephrology, Zhong Da Hospital, Southeast University School of Medicine, Nanjing City, Jiangsu Province, China.
2. Centre for Nephrology, University College London (UCL) Medical School, Royal Free Campus, UK.
Abstract
Background Chronic inflammation plays a crucial role in the progression of cardiac fibrosis. This study investigated whether inflammation exacerbated the progression of cardiac fibrosis in high-fat-fed apolipoprotein E knockout (ApoE KO) mice via endothelial-mesenchymal transition (EndMT).
Methods Twenty-four male ApoE KO mice were divided into normal chow diet (Control), high-fat diet (HFD), or high-fat diet plus 10% casein injection (inflamed) groups for 8 weeks. The body weight of ApoE KO mice was measured at each week. The lipid profile and serum amyloid A (SAA) levels were examined using clinical biochemistry and enzyme-linked immunosorbent assays, respectively. Cardiac lipid and collagen accumulation was visualised with haematoxylin-eosin (HE) and Masson's trichrome staining. EndMT-related molecule expression was examined by immunohistochemistry and Western blotting.
Results SAA levels were increased in the inflamed group compared with the HFD and control groups, suggesting that inflammation was successfully induced. There were no differences in body weight among three groups at each week. Interestingly, inflammation significantly reduced serum total cholesterol, triglyceride, and low-density lipoprotein (LDL) levels compared with the HFD mice. However, both foam cell formation in cardiac blood vessels and cardiac collagen deposition were increased in the inflamed group, as demonstrated by HE and Masson trichrome staining. Furthermore, inflammation reduced protein expression of CD31 and increased protein expression of alpha-smooth muscle actin (α-SMA) and collagen I, which contribute to cardiac EndMT.
Conclusions Inflammatory stress exacerbates the progression of cardiac fibrosis in high-fat-fed ApoE KO mice via EndMT, suggesting that hyperlipidaemia and inflammation act synergistically to redistribute plasma lipids to cardiac tissues and accelerate the progression of cardiac fibrosis.
Keywords: Dyslipidaemia, inflammation, EndMT, cardiac fibrosis
Introduction
Cardiac fibrosis is a common feature in patients with advanced cardiac failure regardless of the aetiology of cardiomyopathy [1]. Cardiac fibrosis, which is associated with decreased microvasculature and the disruption of normal myocardial structure, results from an excessive deposition of extracellular matrix, which is mediated by the recruitment of fibroblasts. However, the origin of these fibroblasts in the adult heart is unclear [2]. Traditionally, adult fibroblasts are derived directly from embryonic mesenchymal cells and increase in number only because of the proliferation of resident fibroblasts. Recent studies have suggested that during fibrosis, bone marrow-derived fibroblasts and endothelial cells contribute to fibroblast accumulation through endothelial-mesenchymal transition (EndMT) in addition to the proliferation of resident fibroblasts [3, 4]. During EndMT, resident endothelial cells delaminate from an organised cell layer and invade the underlying tissue. This mesenchymal phenotype is characterised by reduced expression of endothelial markers, such as CD31 or vascular endothelial cadherin, and increased expression of mesenchymal markers, such as fibroblast-specific protein-1, collagen I, or α-smooth muscle actin (α-SMA), as well as the loss of cell-cell junctions and the acquisition of invasive and migratory properties. Thus, EndMT-derived cells function as fibroblasts in damaged tissue and may have an important role in tissue remodelling and fibrosis [5-8].
Chronic inflammation and hyperlipidaemia are recognised as two risk factors for cardiac fibrosis [9, 10]. Recent studies demonstrated that inflammation and dyslipidaemia act synergistically in organ injuries [11, 12]. Ruan and colleagues demonstrated that inflammatory stress exacerbated lipid accumulation via disruption of LDL receptor (LDLr) feedback regulation in the aorta, liver or kidney [13-15]. However, the synergistic mechanism of inflammation and dyslipidaemia in heart damage has not been defined. The present study sought to explore whether inflammation exacerbates the progression of cardiac fibrosis in high-fat-fed apolipoprotein E knockout (ApoE KO) mice via EndMT.
Materials and methods
Animal Model
Male ApoE KO mice on a C57BL/6 genetic background were studied using protocols that were approved by the Southeast University Committee on the Ethics of Animal Experiments.
Eight-week-old ApoE KO mice were fed normal chow (control group, n=8), a high-fat Western diet containing 21% fat and 0.15% cholesterol (HFD group, n=8), or a high-fat diet plus daily subcutaneous injections of 0.5 mL 10% casein (inflamed group, n=8). The body weight of mice was measured by electron balance at each week. After 8 weeks, blood samples were taken to assess serum amyloid A (SAA) levels and serum lipid profiles (total cholesterol, triglyceride, LDL and HDL), and heart samples were used for histology.
Enzyme-Linked Immunosorbent Assay
SAA serum levels were assessed with kits (Invitrogen, USA).
Lipid profile assessment
The serum lipid profile in ApoE KO mice (total cholesterol, triglycerides, LDL and HDL) was measured at the Zhong Da Hospital's Clinical Biochemistry Department laboratory.
Tissue processing
Heart sections were rinsed with saline and placed in 10% buffered formalin. After treatment, representative sections were embedded in paraffin.
Haematoxylin-eosin (HE) Staining
Paraffin-embedded hearts were sectioned and dewaxed. Sections were stained for 15 minutes with haematoxylin, followed by 1% eosin for 3 minutes. After dehydration, resinene was used to seal the sections to transparency. The sections were observed under light microscopy (×400).
Masson's trichrome staining
Paraffin-embedded hearts (3 μm) were sliced and dewaxed. Myocardial fibrosis was assessed with Masson's trichrome stain. Results were observed under light microscopy (×100).
Immunohistochemistry
Paraffin-embedded sections (4 μm) were subjected to immunohistochemistry. After deparaffinisation, sections were placed in citrate-buffered solution (pH 6.0) and heated for antigen retrieval. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide treatment, and nonspecific binding was blocked with 10% goat serum. Sections were incubated with rabbit anti-mouse primary collagen I antibodies (Abcam, UK) overnight at 4℃, followed by incubation with biotinylated secondary antibodies. Finally, diaminobenzidine tetrahydrochloride substrate was used for the colour reaction. Staining was observed by light microscopy (×100).
Western blot
Equal amounts of total protein from ApoE KO mouse heart homogenates was separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). After transfer, membranes were blocked with blocking buffer for 1 hour at room temperature. Membranes were then incubated with mouse anti-mouse or rabbit anti-mouse CD31, α-SMA, or collagen I primary antibodies (Abcam, UK) overnight at 4℃, followed by incubation with horseradish peroxidase-labelled secondary antibodies for 2 hours. Finally, signals were detected using enhanced chemiluminescence (Amersham Biosciences, UK).
Statistical analysis
All of the data are expressed as the mean ± standard deviation (SD) and were processed with SPSS software 13.0. A one-Way ANOVA was used to compare continuous variables among the three groups where appropriate. Differences were considered to be significant if the P value was less than 0.05.
Results
ApoE KO mouse serum SAA level analysis with or without casein injection
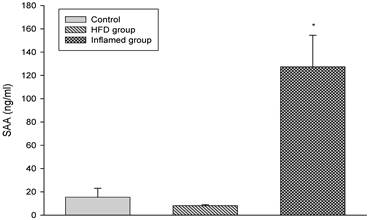
We induced inflammatory atherosclerosis in ApoE KO mice by injecting them with 10% casein. We found that serum SAA was significantly increased in the casein-injected mice compared with the controls, suggesting that systemic inflammation was successfully induced (Figure 1).
There were no differences in body weight change among three groups at each week
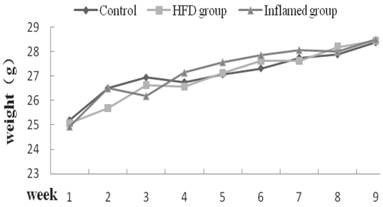
To exclude the effect of body weight on serum levels of total cholesterol, triglyceride LDL, and HDL, we measured the body weight of ApoE KO mice, and found that there were no differences in body weight change at each week among three groups (Figure 2).
Chronic inflammation decreased serum lipid levels compared with the HFD mice
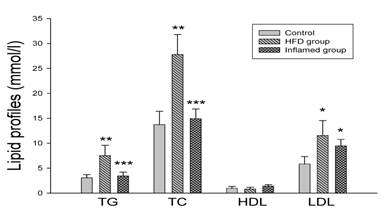
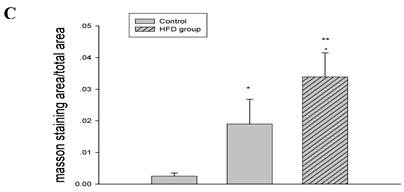
As shown in Figure 3, there were significantly increased serum total cholesterol, triglyceride and LDL levels in high-fat-fed ApoE KO mice compared with the controls. However, serum total cholesterol, triglyceride and LDL levels were decreased in the casein-injected ApoE KO mice compared with the HFD mice (Figure 3).
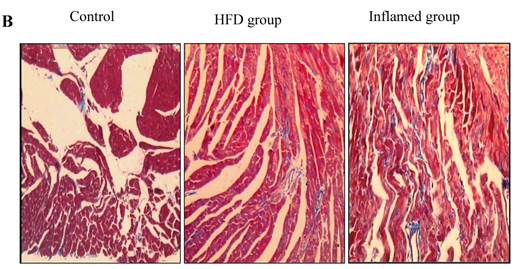
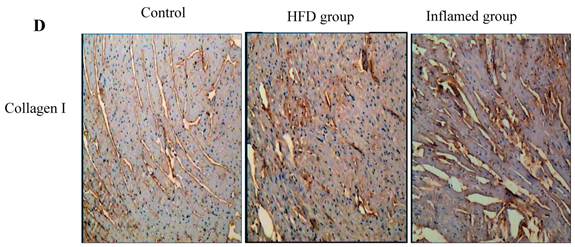
Inflammation increased foam cell formation in cardiac blood vessels and exacerbated cardiac fibrosis in high-fat-fed ApoE KO mice
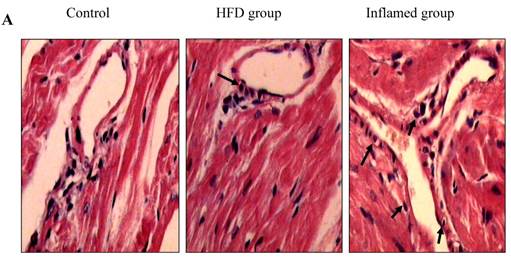
Using HE and Masson's trichrome staining, we determined that the high-fat diet induced foam cell formation in cardiac blood vessels (Figure 4A) and cardiac collagen deposition compared with the control (Figure 4B, 4C). Meanwhile, immunohistochemistry demonstrated that collagen I protein expression was significantly increased in the HFD and inflamed groups compared with the control (Figure 4D). Interestingly, although the serum total cholesterol, triglyceride, and LDL levels in the inflamed group were lower than in the HFD group, the degree of foam cell formation and cardiac fibrosis in the inflamed group was enhanced compared with the HFD group.
Serum SAA levels at experimental termination (n=8). ApoE KO mice were fed normal chow (Control) or Western diet for eight weeks without (HFD group) or with 10% casein injections (Inflamed group). Abbreviations: SAA, serum amyloid A. *p<0.001 vs. Control.
Body weight change at each week (n=8). ApoE KO mice were fed normal chow (Control) or Western diet for eight weeks without (HFD group) or with 10% casein injections (Inflamed group). The body weight change was measured at each week.
Serum lipid profile analysis at experimental termination (n=8). ApoE KO mice were fed normal chow (Control) or Western diet for eight weeks without (HFD group) or with 10% casein injections (Inflamed group). Abbreviations: TC, total cholesterol; TG, triglyceride; LDL, low-density lipoprotein; HDL, high-density lipoprotein; *p<0.05 vs. Control, **p<0.001 vs. Control, ***p<0.001 vs. HFD group.
Effect of inflammation on lipid deposition in cardiac blood vessels and cardiac collagen deposition in high-fat-fed ApoE KO mice (n=8). ApoE KO mice were fed normal chow (Control) or Western diet for eight weeks without (HFD group) or with 10% casein injections (Inflamed group). Cardiac blood vessel lipid deposition was assessed by HE staining (A, original magnification×400), and cardiac collagen deposition was assessed by Masson's trichrome (B, blue colour, original magnification×100) and the values of semiquantitative analysis for the positive areas are expressed as the Mean ±SD from six ApoE KO mice at each group. *p < 0.001 vs. control, **p<0.01 vs. HFD group (C). The collagen I protein expression was measured by immunohistochemical staining (D, brown colour, original magnification×100).
Inflammation accelerated hyperlipidaemia-mediated cardiac fibrosis by contributing to EndMT (n=8). ApoE KO mice were fed normal chow (Control) or Western diet for eight weeks without (HFD group) or with 10% casein injections (Inflamed group). Collagen I, CD31, and α-SMA protein expression was assessed by Western blot (A and B). The histogram represented Mean ± SD of the densitometric scans of the protein bands from eight ApoE KO mice per group, normalized by comparison with β-actin .*p<0.01 vs. control, **p<0.001 vs. control, ***p<0.001 vs. HFD group.
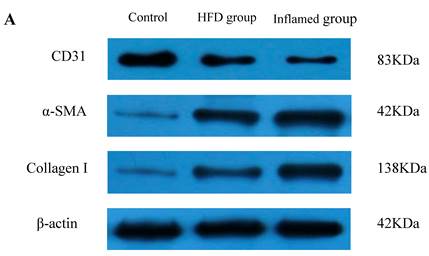
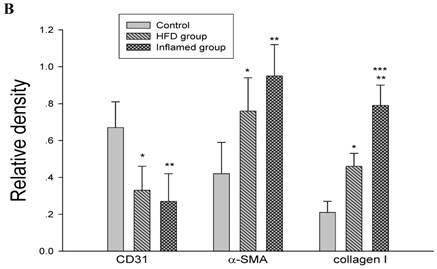
Inflammation accelerated lipid-mediated cardiac fibrosis by contributing to EndMT
To assess the possible mechanisms of hyperlipidaemia and inflammation-induced cardiac fibrosis, we evaluated the effects of inflammation on the protein expression of EndMT biomarkers collagen I, CD31 and α-SMA in the ApoE KO mouse hearts. Western blot demonstrated that inflammation significantly increased the expression of collagen I and α-SMA but decreased CD31 protein expression in high-fat-fed ApoE KO mice (Fig 5A and 5B). These data suggest that inflammation exacerbates the progression of dyslipidemia-induced cardiac fibrosis via EndMT.
Discussion
Cardiac fibrosis is a multi-factorial disease that occurs in several pathological processes, including hypertension, diabetes, radiation, viral myocarditis, and genetic mutations [16]. Inflammatory stress and dyslipidaemia synergise to cause organ injuries [13-15, 17]. Thus, this study investigated the role of both inflammatory stress and dyslipidaemia in cardiac fibrosis, as well as possible mechanisms for their synergy, using ApoE KO mice.
We subcutaneously injected the high-fat-fed ApoE KO mice with 10% casein and successfully induced inflammatory atherosclerosis, as shown by significantly increased plasma SAA levels compared with the HFD group. These results demonstrated that inflammatory stress markedly exacerbated lipid accumulation in cardiac blood vessels and cardiac collagen deposition, which contributed to the progression of cardiac fibrosis. Interestingly, serum total cholesterol, triglyceride and LDL levels in the casein-injected mice were lower than in the HFD group. In order to exclude the factors from reduced food intake or increased fecal excretion, we checked the body weight in three groups of mice. As shown by Figure 2, there were no differences about body weight change at each week among three groups. The results suggest that the balance of food intake and fecal excretion in casein injection group was roughly equivalent to that in the control and HFD group. Our previous in vivo and in vitro studies demonstrated that inflammatory stress disrupted cholesterol homeostasis through dysregulation of the LDLr pathway and exacerbated the progression of atherosclerosis, vascular calcification, and liver injury [14, 18, 19]. Therefore, inflammatory stress may induce lipid redistribution from the circulation to the cardiac tissues, thereby causing hypolipidaemia and cardiac fibrosis [20].
To explore potential mechanisms, we further assessed the effects of dyslipidaemia and inflammatory stress on EndMT in ApoE KO mouse hearts. Our results demonstrated that hyperlipidaemia upregulated the protein expression of collagen I and α-SMA but decreased the expression of CD31, interestingly, which were deteriorated by inflammation. These data suggest that inflammatory stress and hyperlipidaemia in combination contribute to the progression of cardiac fibrosis via EndMT.
Recently, other studies reported that inflammatory stress plays an essential role in cardiac fibrosis. Alvarez et al. demonstrated that autoimmune-associated congenital heart blockage might result from pathogenic cross-talk between cardiac inflammatory and profibrotic pathways. Immune complexes stimulated fibrosis in human foetal cardiac fibroblasts through Toll-like receptor 7 [21]. Clinical studies demonstrated that myocardial tissue from patients with cardiac fibrosis exhibited the activation of nuclear factor-kappa B (NF-κB) and increased NF-κB-regulated gene expression [22, 23]. In addition to cardiac fibrosis, Chaudhuri demonstrated that long-term exposure to tumour necrosis factor (TNF-α) or interleukin-1β (IL-1β) induced permanent transformation of human epithelioid dermal microvascular endothelial cells into myofibroblasts in cell culture, demonstrating that EndMT might be involved in skin fibrogenesis [24]. Yamauchi reported that TNF-α induced EndMT in lung carcinoma via TGF-β signalling [25]. Our previous study also demonstrated that high glucose induced endothelial damage via an angiotensin II-mediated EndMT, and subsequently caused cardiovascular events in diabetes [26].
Dyslipidaemia was also strongly associated with the progression of cardiac fibrosis [9]. Qin et al. demonstrated that simvastatin inhibited cardiac hypertrophy and fibrosis by reducing total cholesterol and matrix metalloproteinase-9 levels in Western diet-fed Apolipoprotein E-deficient mice [27]. Wendt et al. suggested a link between hypercholesterolemia and left ventricular fibrosis in Dahl salt-sensitive rats, which are genetically modulated by chromosome 19 [28].
In summary, our findings demonstrated for the first time that inflammatory stress exacerbates the progression of cardiac fibrosis in high-fat-fed ApoE KO mice via EndMT, suggesting that hyperlipidaemia and inflammation synergise to redistribute lipid from the plasma to the heart, thereby accelerating the progression of cardiac fibrosis.
Acknowledgements
This work was supported by grants 81070571 and 81170792 from the National Natural Science Foundation of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vasan RS. Diastolic heart failure. BMJ. 2003;327:1181-2
2. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E. et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature medicine. 2007;13:952-61
3. Rivera-Feliciano J, Lee KH, Kong SW, Rajagopal S, Ma Q, Springer Z. et al. Development of heart valves requires Gata4 expression in endothelial-derived cells. Development. 2006;133:3607-18
4. Zhou B, von Gise A, Ma Q, Hu YW, Pu WT. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Developmental biology. 2010;338:251-61
5. Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circulation research. 2004;95:459-70
6. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer research. 2007;67:10123-8
7. Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. American journal of physiology Lung cellular and molecular physiology. 2007;293:L1-8
8. Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N. et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407-18
9. Klaus JR, Hurwitz BE, Llabre MM, Skyler JS, Goldberg RB, Marks JB. et al. Central obesity and insulin resistance in the cardiometabolic syndrome: pathways to preclinical cardiovascular structure and function. Journal of the cardiometabolic syndrome. 2009;4:63-71
10. Kalogeropoulos AP, Georgiopoulou VV, Butler J. From risk factors to structural heart disease: the role of inflammation. Heart failure clinics. 2012;8:113-23
11. Yaoita H, Yoshinari K, Maehara K, Sando M, Watanabe K, Maruyama Y. Different effects of a high-cholesterol diet on ischemic cardiac dysfunction and remodeling induced by coronary stenosis and coronary occlusion. Journal of the American College of Cardiology. 2005;45:2078-87
12. Spillmann F, Van Linthout S, Tschope C. Cardiac effects of HDL and its components on diabetic cardiomyopathy. Endocrine, metabolic & immune disorders drug targets. 2012;12:132-47
13. Ruan XZ, Moorhead JF, Tao JL, Ma KL, Wheeler DC, Powis SH. et al. Mechanisms of dysregulation of low-density lipoprotein receptor expression in vascular smooth muscle cells by inflammatory cytokines. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1150-5
14. Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead JF, Varghese Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology. 2008;48:770-81
15. Xu ZE, Chen Y, Huang A, Varghese Z, Moorhead JF, Yan F. et al. Inflammatory stress exacerbates lipid-mediated renal injury in ApoE/CD36/SRA triple knockout mice. Am J Physiol Renal Physiol. 2011;301:F713-22
16. Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiological reviews. 1999;79:215-62
17. Steinberg D. Hypercholesterolemia and inflammation in atherogenesis: two sides of the same coin. Molecular nutrition & food research. 2005;49:995-8
18. Liu J, Ma KL, Gao M, Wang CX, Ni J, Zhang Y. et al. Inflammation Disrupts the LDL Receptor Pathway and Accelerates the Progression of Vascular Calcification in ESRD Patients. PloS one. 2012;7:e47217
19. Ma KL, Ruan XZ, Powis SH, Moorhead JF, Varghese Z. Anti-atherosclerotic effects of sirolimus on human vascular smooth muscle cells. American journal of physiology Heart and circulatory physiology. 2007;292:H2721-8
20. Ruan XZ, Varghese Z, Moorhead JF. An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol. 2009;5:713-21
21. Alvarez D, Briassouli P, Clancy RM, Zavadil J, Reed JH, Abellar RG. et al. A novel role of endothelin-1 in linking Toll-like receptor 7-mediated inflammation to fibrosis in congenital heart block. The Journal of biological chemistry. 2011;286:30444-54
22. Arkan MC, Greten FR. IKK- and NF-kappaB-mediated functions in carcinogenesis. Current topics in microbiology and immunology. 2011;349:159-69
23. Lorenzo O, Picatoste B, Ares-Carrasco S, Ramirez E, Egido J, Tunon J. Potential role of nuclear factor kappaB in diabetic cardiomyopathy. Mediators of inflammation. 2011;2011:652097
24. Chaudhuri V, Zhou L, Karasek M. Inflammatory cytokines induce the transformation of human dermal microvascular endothelial cells into myofibroblasts: a potential role in skin fibrogenesis. Journal of cutaneous pathology. 2007;34:146-53
25. Yamauchi Y, Kohyama T, Takizawa H, Kamitani S, Desaki M, Takami K. et al. Tumor necrosis factor-alpha enhances both epithelial-mesenchymal transition and cell contraction induced in A549 human alveolar epithelial cells by transforming growth factor-beta1. Experimental lung research. 2010;36:12-24
26. Tang R, Li Q, Lv L, Dai H, Zheng M, Ma KL. et al. Angiotensin II mediates the high-glucose-induced endothelial-to-mesenchymal transition in human aortic endothelial cells. Cardiovascular diabetology. 2010;9:31
27. Qin YW, Ye P, He JQ, Sheng L, Wang LY, Du J. Simvastatin inhibited cardiac hypertrophy and fibrosis in apolipoprotein E-deficient mice fed a "Western-style diet" by increasing PPAR alpha and gamma expression and reducing TC, MMP-9, and Cat S levels. Acta pharmacologica Sinica. 2010;31:1350-8
28. Wendt N, Schulz A, Qadri F, Bolbrinker J, Kossmehl P, Winkler K. et al. Genetic analysis of salt-sensitive hypertension in Dahl rats reveals a link between cardiac fibrosis and high cholesterol. Cardiovascular research. 2009;81:618-26
Author contact
![]() Corresponding author: Bi Cheng Liu. Institute of Nephrology, Zhong Da Hospital, Southeast University School of Medicine, NO.87, Ding Jia Qiao Road, Nanjing City, Jiangsu Province, China, 210009. Tel: 0086 25 83285139 Fax 0086 25 83285130, E-mail: liubc64com.cn
Corresponding author: Bi Cheng Liu. Institute of Nephrology, Zhong Da Hospital, Southeast University School of Medicine, NO.87, Ding Jia Qiao Road, Nanjing City, Jiangsu Province, China, 210009. Tel: 0086 25 83285139 Fax 0086 25 83285130, E-mail: liubc64com.cn
Received 2012-12-17
Accepted 2013-2-25
Published 2013-2-28