Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(4):362-376. doi:10.7150/ijms.5698 This issue Cite
Research Paper
Improved Culture Conditions for the Growth and Detection of Borrelia from Human Serum
Eva Sapi1,2 ![]() , Namrata Pabbati1, Akshita Datar1, Ellen M Davies1, Amy Rattelle1, Bruce A Kuo1
, Namrata Pabbati1, Akshita Datar1, Ellen M Davies1, Amy Rattelle1, Bruce A Kuo1
1. Research Division of Advanced Laboratory Services Philadelphia PA, USA;
2. Department of Biology and Environmental Science, University of New Haven, West Haven CT, USA.
Received 2012-12-12; Accepted 2013-2-11; Published 2013-2-18
Abstract
In this report we present a method to cultivate Borrelia spirochetes from human serum samples with high efficiency. This method incorporates improved sample collection, optimization of culture media and use of matrix protein. The method was first optimized utilizing Borrelia laboratory strains, and later by demonstrating growth of Borrelia from sera from fifty seropositive Lyme disease patients followed by another cohort of 72 Lyme disease patients, all of whom satisfied the strict CDC surveillance case definition for Lyme disease. The procedure resulted in positive cultures in 47% at 6 days and 94% at week 16. Negative controls included 48 cases. The positive identification of Borrelia was performed by immunostaining, PCR, and direct DNA sequencing.
Keywords: Clinical microbiology, Vector-borne diseases, Lyme disease, Spirochetes.
Introduction
It has long been an elusive goal to develop a successful in vitro culture method for wild-type, pathogenic Borrelia. Multiple attempts using a variety of methods have not been able to obtain a high yield or keep them alive for an extended period of time [1] [2]. A successful culture system would be highly useful, especially in light of the recognized public health impact of the pathogen Borrelia burgdorferi, the causative agent of Lyme disease.
Lyme disease is a rapidly emerging infectious disease in the United States [3]. In most cases of Lyme disease, early detection and treatment with a course of antibiotics results in a successful outcome [2], [4]. However, because the signs and symptoms of disseminated Lyme disease are shared with many other diseases [5], and because currently recommended diagnostic laboratory tests are not 100% sensitive, many cases of Lyme disease go undiagnosed and untreated, putting an infected patient at risk for developing a debilitating long-term illness [1] [6]-[9]. The current recommended standard for Lyme disease testing employs an indirect 2-step serological assay that detects host immune reactivity to the Lyme disease bacteria [1] [2] [5]. In a significant number of cases the serologic assays are not sufficiently sensitive or specific [1], [10]-[15] in part due to the presence of immune suppressive and modulating agents transmitted by the tick and secreted by the Borrelia spirochete [13]-[15] and antigen-antibody complex formation [16]-[19]. Thus DNA-detection methods (polymerase chain reaction or PCR) have been developed but the assay has less than optimal sensitivity as the concentration of Borrelia spirochetes in blood, plasma, or serum samples from Lyme disease patients can be very low [1], [2], [20]-[23]. In addition, inhibitors of PCR have been demonstrated to exist in the peripheral blood [24]. For technical reasons false-positive PCRs remain a concern [22], [25]. Therefore, there is a need to improve methods to detect Borrelia infection in patients including methods to cultivate Borrelia from human blood, urine, or tissue samples [26].
The Borrelia-specific Barbour-Stoenner-Kelly media (BSK media) has been used to culture Borrelia [27], but the sensitivity is suboptimal [1], [2], [10], [28]-[30]. The reported sensitivities range from as low as 5% to a maximum of 88% in skin samples but a maximum of only 40-44% for peripheral blood samples [1], [10], [28]-[32]. Because of these difficulties, Borrelia cultures until now have not been widely used.
We report an improved method to cultivate Borrelia from human blood samples with a success rate of 94 %. This culture method incorporates two-step process that consists of a short-term starter culture that is used to seed a long-term phase, each utilizing different cultivation environments. Combining the two phases allows for more rapid initial growth and ultimately a higher yield than has previously been reported. This highly sensitive and specific method to detect live Borrelia cultivated from patient's serum will improve our ability to make the diagnosis in cases in which serologic or PCR based assays are not informative. In addition, cultures have been maintained for as long as eight months, offering the potential to serve as a useful source of clinical isolates for further study.
Materials and Methods
Abbreviations/nomenclature
Members of the Borrelia genus often referred to as Borrelia burgdorferi sensu lato, Borrelia species, Borrelia spp, will be referred to in this study by the genus name, Borrelia. When species of Borrelia are referred to here, they will be indicated by the specific species name.
Culture of established cell lines
Low passage isolates of two different Borrelia burgdorferi strains, B31 (ATCC # 35210) and 297 (ATCC # 53899), Borrelia afzelii (ATCC # 51992), and Borrelia garinii (ATCC # 51991) were obtained from American Type Tissue Collection. Escherichia coli DH5α strain was purchased from Life Technologies. Borrelia strains were maintained in complete BSK-H media, which includes 6% rabbit serum (complete media from Sigma-Aldrich #B8291) at 34oC with 5% CO2 and 100% humidity. Each new lot of complete BSK-H media was tested in a reference strain culture as a measure of quality assurance. Escherichia coli cells were maintained in standard Luria Broth media (Sigma) at 37oC in a shaking incubator.
Blood collection
Full informed consent was obtained from each study participant and HIPAA-compliant privacy measures were maintained. All patients who participated in this study had not been exposed to antibiotics for a minimum of 4 weeks prior to blood sample collection [33] [34]. Sera from 50 seropositive patients were used to establish the culture media and conditions. After the cultivation parameters were established, sera from an additional 72 Lyme disease patients (26 males and 46 females ranging in age from 3 to 80 years with average of 42 years) were collected between March 2011 and May 2012 and used to evaluate this culture method. All of these patients had a clinical presentation consistent with Lyme disease and their illness was confirmed by laboratory evidence of infection using the Center for Disease Control (CDC) recommended, 2-tiered serological method, thus they all satisfied the strict CDC surveillance case definition for Lyme disease.
To determine the specificity of the test we collected serum from 48 healthy controls that had no history of either tick-bites or Lyme disease (21 males and 27 females ranging in age from 27 to 71 years with an average of 43 years). Patients and controls were selected from private medical practices from 15 different States (CA, AR, CT, NH, NJ, NY, MA, MD, MN, OR, PA, TN, UT, VT). Samples were delivered to the laboratory and cultures were initiated within 24-30 hours of collection.
Culture protocol
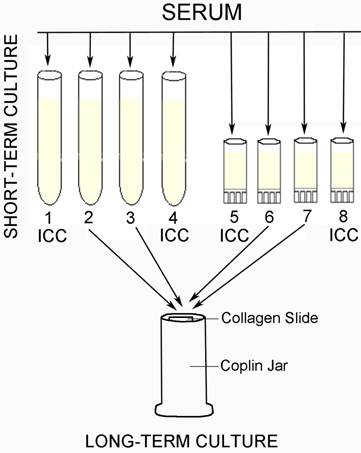
Blood samples (~30 ml /patient) were collected in the following tubes: 9.0 ml peripheral blood in a 15.0 ml polypropylene Falcon tube containing 5 ml BSK-H, two samples consisting of 9.0 ml peripheral blood in 10.0 ml red top (no additive) Vacutainer tube, or Vacutainer tubes containing EDTA (purple top). Blood samples were transported to the laboratory overnight at room temperature and upon receipt they were allowed to sit undisturbed for several hours at room temperature to allow serum separation. The serum was divided into eight starter cultures for each clinical sample and mixed with modified BSK-H media (mBSK-H) - four in the standard 15 ml glass tube (13 ml final volume) and four in the small 2ml cryo tubes (1.8 ml final volume) as summarized in Table 2. Cultures were incubated at 34º C with 5% CO2 and 100% humidity. The lids of all culture tubes were closed loosely to allow limited gas exchange between the culture medium and the environment. Starter cultures were harvested after 6 days and used for either immunocytochemical studies or to seed a long-term culture. Long-term cultures were established by combining and directly transferring cells and media from culture tubes to a Coplin jar with an additional 15 ml mBSK-H media (total volume of 34 ml), a 5 µg rifampicin disc (BD Scientific) and 2 collagen coated slides (Rat-tail type I; BD Scientific) and maintained through 16 weeks at 34oC with 5% CO2 and 100% humidity. After 8 weeks, one slide was checked under dark field microscopy and used for polyclonal anti-Borrelia antibody detection analysis. If the fluorescent antibody result was negative at 8 weeks of culture, another collagen coated slide was placed in the Coplin jar and half of the culture media (~17 ml) was replaced with fresh mBSK-H medium and the culture was incubated for an additional 8 weeks.
Description of the different culture environments for short-term cultures. * = modified BSK-H (including 12 % rabbit serum, 100 µg/ml of DTT and rifampicin as antibiotics). ICC=Immunocytochemistry, LTC=Long-term Culture.
| Tube# | Tube type | Volume of the culture medium* | Seed source | Day 6 use |
|---|---|---|---|---|
| 1 | 15 ml | 11.0 ml | 2.0 ml serum/BSK-H | ICC |
| 2 | 15 ml | 11.0 ml | 2.0 ml serum/BSK-H | LTC |
| 3 | 15 ml | 11.5 ml | 1.5 ml serum | LTC |
| 4 | 15 ml | 11.5 ml | 1.5 ml serum | ICC |
| 5 | 2 ml | 1.4 ml | 0.4 ml serum/BSK-H | ICC |
| 6 | 2 ml | 1.4 ml | 0.4 ml serum/BSK-H | LTC |
| 7 | 2 ml | 1.6 ml | 0.2 ml serum | LTC |
| 8 | 2 ml | 1.6 ml | 0.2 ml serum | ICC |
Immunodetection with polyclonal and monoclonal antibodies
For detection of Borrelia using the polyclonal antibody, cells from the starter culture tubes (Table 1) were centrifuged at 9,600xg for 10 minutes at room temperature, washed with 1x PBS buffer, pH 7.4, and centrifuged again at 9,600xg. The pellet was resuspended in 75 µl of 1x PBS buffer and spread on a microscope slide (SuperFrost+, Fischer Scientific). Cells were fixed by incubating the slides in acetone for 10 minutes at -20 ºC. For collagen-coated slides, the cells were washed twice with 1x PBS, fixed in ice-cold acetone:methanol (1:1) mixture for 10 minutes at -20 ºC, dried for 10 minutes at room temperature in a laminar air-flow hood, and further dried at 37º for 20 minutes. 100 µl of polyclonal antibody (PA-1-73005 polyclonal FITC labeled anti-Borrelia burgdorferi antibody, Thermo Scientific, diluted 1:50 in 1% BSA/1x PBS) was added to the dried and fixed cells (300 µl for collagen-coated slides). For a negative control, the primary antibody was omitted and replaced with normal rabbit serum. The slides were incubated at 37ºC in a humidified chamber for 1 hour, washed with 1x PBS for 5 minutes at room temperature, then rinsed twice in double distilled water and allowed to dry in the laminar air-flow hood for 10 minutes. The slides were mounted with Vectashield mounting medium with DAPI counterstain (Vector Labs) and images were taken by fluorescent microscopy. Unless noted otherwise, microscopy was performed at 400x magnification using an Olympus BX40 fluorescent microscope with attached digital camera. An internal validation study for the specificity of the polyclonal antibody showed that it is able to recognize reference cultures of Borrelia burgdorferi, Borrelia afzelii and Borrelia garinii, but not Escherichia coli or Treponema denticola (data not shown).
Summary of the positive Borrelia clinical samples cultured in 15ml and 2ml tubes tubes/collection tubes. * positive samples grown in small tubes only.
| POSITIVE CULTURES | CULTURE TUBES | |
|---|---|---|
| 15ml | 2ml | |
| 1 | + | - |
| 2 | - | +* |
| 3 | + | - |
| 4 | + | - |
| 5 | - | +* |
| 6 | + | - |
| 7 | + | + |
| 8 | + | + |
| 9 | - | +* |
| 10 | + | + |
| 11 | + | - |
| 12 | + | - |
| 13 | + | - |
| 14 | + | - |
| 15 | + | - |
| 16 | + | - |
| 17 | + | - |
| 18 | + | + |
| 19 | - | +* |
| 20 | + | - |
| 21 | - | +* |
| 22 | + | - |
| 23 | + | - |
| 24 | + | - |
| 25 | + | - |
| 26 | + | - |
| 27 | + | + |
| Total | 22 | 10 |
For monoclonal antibody detection of Borrelia, cells were collected and adhered to a microscope slide as described for the polyclonal antibody protocol. The cells were fixed by incubation in fresh 4% paraformaldehyde solution for 20 minutes at room temperature followed by incubation in cold methanol for 20 minutes at -20 ºC. The cells were treated in 0.3% Triton X-100 solution for 15 minutes at 37 ºC, then washed twice with 1x PBS, twice with double distilled water, air dried for 10 minutes in a laminar air-flow hood, and further dried at 37 ºC for 20 minutes. The slides were blocked with 100 µl diluted normal goat serum (5% in 1% BSA/1x PBS, Vector Labs) in a humidified chamber at 37 ºC for 20 minutes. The slides were washed twice with 1x PBS, and twice with double distilled water. 100 µl of monoclonal antibody (MA-1-7006, Thermo Scientific, raised against Bb, diluted 1:10 in 1% BSA/1x PBS) was added to each slide and incubated in a humidified chamber at room temperature overnight (~16h). For a negative control, the monoclonal antibody was omitted and replaced with normal mouse serum. The slides were washed twice with 1xPBS, and twice with distilled water. 100 µl of goat anti-mouse secondary antibody (DyLight 488 fluorescent dye labeled, diluted 1:200 in 1% BSA/1xPBS) was added to each slide and incubated in a humidified chamber at room temperature for 1 hour under reduced light conditions. The slides were washed twice with 1x PBS, and twice with double distilled water and air dried in the dark under a laminar flow hood for 15 minutes and counterstained with Vectashield with DAPI (Vector Labs). Images were acquired by fluorescent microscopy as described above. An internal validation study for the specificity of this monoclonal antibody showed that it is able to recognize Borrelia burgdorferi B31 and 297 strains but not the reference strains of Borrelia afzelii and Borrelia garinii, Escherichia coli or Treponema denticola (data not shown).
DNA preparation from patient samples and cultures
DNA was prepared from a portion of the initial patient serum sample; the remainder of the serum sample was used to seed the starter cultures. For analysis at short term, cells were collected immediately from serum (1-4 ml) by centrifugation at 9600xg for 10 minutes. For analysis at 8 or 16 weeks, cells were collected from the long-term culture media (10 ml) or from the second collagen slide using a cell scraper in 1 ml of the culture media, by centrifugation at 9600xg for 10 minutes. DNA was extracted from cell pellets using the QiaAmp DNA extraction kit (Qiagen) according to the manufacturer's instructions. Samples were eluted twice from the QiaAmp column with 50 µl QiaAmp elution buffer, pooled and stored at -20oC until analysis.
PCR reactions
Each positive culture sample was tested by PCR using primers designed to amplify Borrelia 16S ribosomal RNA small subunit or CTP synthase genes. The 16S rRNA gene was amplified in a single reaction using primers F: 5'- CCTGGCTTAGAACTAACG-3'; R: 5'-CCTACAAAGCTTATTCCTCAT-3' in a 50 μl reaction containing HotStarTaq buffer (Qiagen) 1.5 mM MgCl2, 25 pmoles of each primer, and 2.5 units of HotStarTaq DNA polymerase (Qiagen). Reaction conditions were an initial denaturation at 94oC for 15 minutes, followed by 40 cycles of 94oC/30s, 50oC/30s, 72oC/1 min. then a final extension at 72oC/5min.
CTP synthase sequences were amplified in nested reactions as described previously by Margos G et al [35]. First round primers were F: 5'-ATTGCAAGTTCTGAGAATA-3' and R: 5'-CAAACATTACGAGCAAATTC-3' in a 25 μl reaction with HotStarTaq buffer (Qiagen), 25 pmoles of each primer, MgCl2 adjusted to 2.5 mM, and 1.5 units HotStarTaq. Reaction conditions were an initial denaturation at 94oC for 15 minutes, followed by 94oC/30s, a 55oC step down from to 48oC decreasing 1C per cycle, followed by 30 cycles of 94oC/30s, 48oC/30s, 72oC/30s and a final extension at 72oC/5min. The nested reaction primers were F: 5'- GATATGGAAAATATTTTATTTATTG -3' and R: 5'-AAACCAAGACAAATTCCAAG -3' in a 50 µl reaction containing 25 pmoles of each primer, 2.5 mM MgCl2, 1.5 units HotStarTaq. Reaction conditions were an initial denaturation at 94oC for 15 minutes, followed by 35 cycles of 94oC/30s, 50oC/30s, 72oC/30s and a final extension at 72oC/5min. The PCR products were analyzed by standard agarose gel-electrophoresis.
Sequencing and sequence analyses
PCR products were purified using the QIAquick PCR purification kit (Qiagen) according to the manufacturer's instructions. Samples were eluted twice in 30 µl, and the eluates from each sample were pooled and sequenced in both directions using the primers that generated the products. Sequencing reactions were performed by Eurofins/MGW/Operon (Huntsville, AL). Sequence alignment and neighbor-joining phylogenetic analyses were conducted using MEGA version 5 [36]. Tree support was evaluated by bootstrapping with 500 replications.
Nucleotide sequence accession numbers
Nucleotide sequences for CTP synthase locus for 51 isolates have been deposited in GenBank (http://www.ncbi.nlm.nih.gov) with the following sequential accession numbers: JX867374 through JX867424.
Statistical analyses
Statistical analyses were performed using modified Wald method with a 95% confidence interval as well as the two-sample paired t-test utilized using NCSS statistical software (NCSS LLC, Kaysville, UT).
Results
Optimization of BSK-H media
Standard BSK-H medium is the best available medium to cultivate Borrelia. We first attempted to improve the medium by testing a range of constituents including additional serum, reducing agents, and various antibiotics. Bb strains B31 and 297 were cultured in BSK-H medium containing different amounts of rabbit serum (6%, 12% or 20%), and the culture was propagated at 34 °C with 5% CO2 and 100% humidity. The culture was examined for spirochete growth by dark field microscopy on day 6. While all three serum concentrations supported spirochete growth, culturing spirochetes in 12% rabbit serum yielded approximately 25% more spirochetes for both strains (Figure 1, Panel A, p=0.008).
shows the effect of different concentrations of rabbit serum (Panel A), DTT (Panel B) and antibiotic concentrations (Panel C) on the growth of Borrelia burgdorferi B31 strains. The cells were cultured for 6 days with different concentration of rabbit serum, DTT and antibiotics as depicted in the Figure and cellular growth was evaluated by direct cell counting of motile spirochetes using dark field microscopy. Each set of data represents three independent experiments (mean ± SD). Statistical significance was determined using two-sample paired t-test. The data *p-values <0.05 indicates statistical significance as related to control.
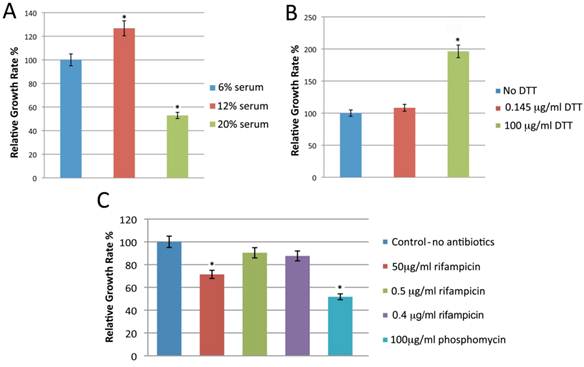
Dithiothreitol (DTT) is a reducing agent that has previously been found to improve the growth of Borrelia in vitro [37]. Bb B31 and 297 strains were cultured in BSK-H medium with 12% rabbit serum and either 0. 145 μg/ml or 100 µg/ml DTT. The spirochete cultures were incubated in a CO2 incubator at 34°C with 5% CO2 and 100% humidity for 6 days. Compared to control or low dose DTT (0.145 μg/ml), 100 µg/ml of DTT nearly doubled the spirochete yield (p=0.0002) for both strains (Figure 1, Panel B).
Antibiotics are commonly added to cultures to suppress the overgrowth of other organisms. To test the effect of this on Borrelia cultures, Bb B31 and 297 strains were cultivated in BSK-H medium containing 12% rabbit serum, 100 μg/ml DTT and one of the following antibiotics: 0.5 µg/ml rifampicin, a 5 µg rifampicin disc (resulting in final concentration of 0.4 µg/ml), 50 µg/ml rifampicin, or phosphomycin. The spirochetes were incubated in a CO2 incubator at 34°C with 5% CO2 and 100% humidity for 6 days and growth was evaluated by dark field microscopy. Culturing spirochetes with either 0.4 or 0.5 µg/ml rifampicin had no effect on spirochete yield, while using a higher concentration (50 µg/ml) of rifampicin significantly decreased the spirochete yield (Figure 1, Panel C, p=0.001). Similarly when three different concentrations of phosphomycin were included in the culture medium, spirochete yield was significantly reduced (data not shown). Therefore we did not include this antibiotic in subsequent experiments.
Use of rifampicin alone (0.5 µg/ml or a 5 µg disc) in the culture prevented contamination in 120 of 122 cases. Thus we utilized BSK-H medium with 12% normal rabbit serum, 100 µg/ml DTT, and either 0.4 or 0.5 µg/ml rifampicin in our subsequent cultures and this will be referred to as modified BSK-H (mBSK-H) medium.
Initial sample collection
50 seropositive Lyme disease patients were studied to test variables in blood collection. Specifically, the use of plasma or serum from the peripheral blood with or without the addition of BSK-H media at the time of collection was examined. Peripheral blood samples were collected in purple-top Vacutainer tubes (EDTA additive, resulting in a plasma sample), in red-top Vacutainer tubes (no additive, resulting in a serum sample), and in 15 ml sterile Falcon tubes with 5 ml of BSK-H medium and were shipped overnight to set up the culture. A positive culture was determined by dark field microscopy and immunocytochemical methods. Collectively, 27 positive cultures were obtained from either the additive-free or BSK-H containing tubes. The tube with whole blood plus BSK-H medium provided the best environment for the 24h transit as this method produced the highest number of positive cultures (24/50, 48%). However, the additive-free tubes (serum) provided a sufficient transit environment for 11 samples (22%) with 8 samples growing from both tube formats (16%). Because 3 cultures grew exclusively in the additive-free tube and to ensure no loss of spirochetes due to transport tube preference, further experiments utilized both transport systems. Blood collected in EDTA-containing tubes gave the lowest yield of 1/50 and thus was abandoned from further use.
Testing different culture tubes
Most previous studies have cultured Borrelia in 15 ml glass tubes which were tightly closed and placed in a standard 33-35 degree Celsius incubator [1], [27]. More recently, a smaller 2 ml polypropylene tube has been found to increase the yield [38]. Therefore, the two different tubes were compared. Serum collected from 50 seropositive Lyme disease patients was inoculated into the two different tubes and cultured with mBSK-H media. Cultures from 27 of these 50 patients were positive for Borrelia by 6 days. Of the 27 positive cultures, 5 were positive in both culture environments while 17 were positive in the 15 ml glass tube only, and 5 samples grew exclusively in the 2 ml tube system as outlined in Table 1. Therefore, to maximize yield, both 15 ml and 2 ml tube formats were included in the final protocol.
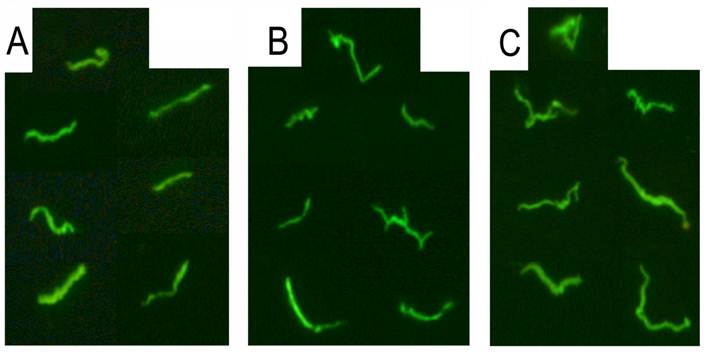
Figure 2 is a representative picture of three successful cultures from seropositive Lyme disease patients after 6 days of culturing. PCR and direct sequencing of the 16S rDNA amplicon further confirmed the presence of Borrelia spirochetes.
shows immunocytochemical images of representative cultures from three seropositive Lyme disease patients (Panel A, B and C). The serum samples were collected and cultured for 6 days and immunostained with polyclonal fluorescent antibodies against Borrelia burgdorferi as described in Materials and Methods. Magnification 400x.
Establishment of Long-term culture environment
Each of the 50 cultures from seropositive Lyme patients described above was continued by adding fresh mBSK-H media and incubating them for additional time. However, after 8-10 days, further growth of the spiral form ceased and spirochetes tended to convert into different morphological forms (data not shown) as described previously [39]-[42]. Multiple attempts to revert them back to spiral form were unsuccessful regardless of the use of several different media such as the original BSK-II medium [27], modified Kelly medium (MKP medium) [43], MPM medium [44], and BSK modified medium [45]. In addition, several solid BSK-based media [46] and PMR-agar media [43] were similarly tested but likewise were not capable of maintaining spirochetal growth.
We thus explored other variables: media and tubes (5-13 ml in 15 ml glass tubes and 1ml-1.8ml in the 2 ml small cryo tubes), other tube sizes (5ml and 10ml tubes), tube material (glass and plastic), culture temperature (28-37 degree Celsius), CO2 concentration (0-5%), and O2 concentration using BD BBL Gaspak systems (anaerobic and semi-anaerobic systems). Despite altering these culture conditions, there was no improvement in growth.
In summary, while the addition of serum, a reducing agent and rifampicin had a positive effect on the short-term Borrelia cultures both by increasing yield and rate of initial growth, additional parameters needed to be optimized to improve long-term Borrelia cultures.
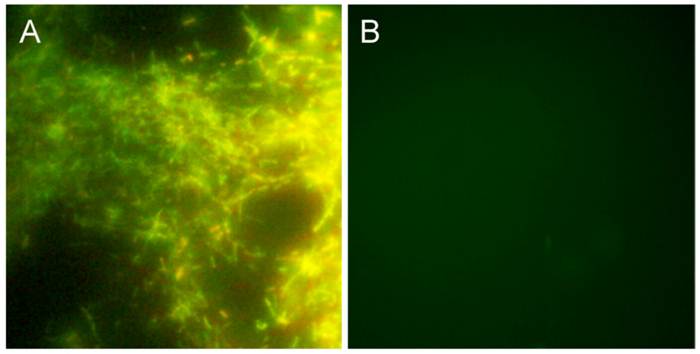
In infected mammals, Borrelia are most often found to be sequestered in collagen and especially collagen type I [47] [48]. A collagen matrix has been shown to support growth of Borrelia burgdorferi B31 and 297 strains in vitro [38] [49]. To study this further we initiated an experiment in which different amounts of Bb B31 cells (1, 10, 100, 1000) were spiked into 10 ml of whole peripheral blood from healthy donors and after a short term culture of 6 days in mBSK-H the culture was transferred to long-term cultures set up in Coplin jars with collagen-coated slides in a total of 45 ml of mBSK-H media. After 14 days, an accumulation of Borrelia growth was observed on all collagen coated slides. To evaluate this growth, a slide was removed from the Coplin jar and stained with the BacLight Live/Dead staining kit (see Materials and Methods). Figure 3A demonstrates a large thriving colony of attached spirochetes, with a very low proportion of non-viable spirochetes. Blood samples that were not spiked with Borrelia cells did not produce any spirochetes on the collagen-coated slides (Figure 3B). These data confirm that under favorable conditions human blood containing as few as one Borrelia B31 spirochete can be cultured in mBSK-H with a collagen source, which supports the original hypothesis that a matrix-supported environment is beneficial for their growth.
shows BacLight DEAD/LIVE stained Borrelia burgdorferi on collagen-coated slides. For this experiment, 1 (Panel A) or 0 (Panel B) bacterial cell was spiked into 10 ml of human blood and cultured in a Coplin jar with collagen-coated slides and mBSK-H culture medium for 4 weeks and stained with Baclight DEAD/LIVE stains as described in Materials and Methods. Green staining=live cells, red staining=dead cells. Magnification: 200X.
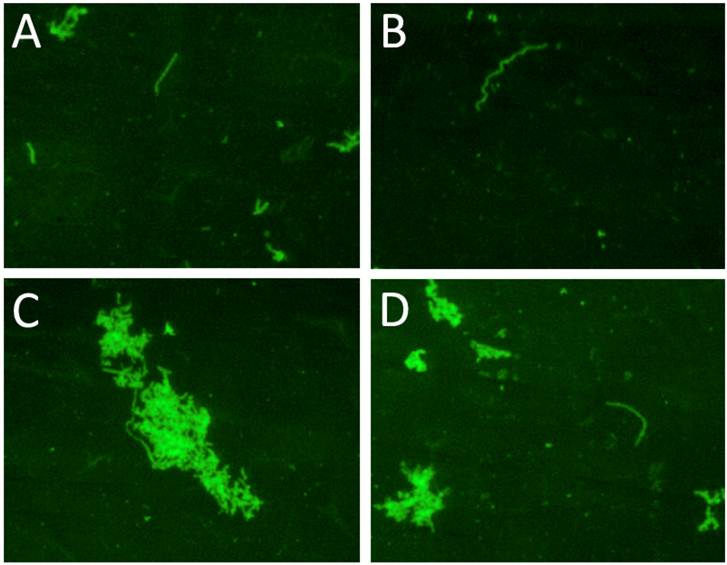
To explore whether a collagen source would be beneficial to growth of wild type Borrelia, 10 cultures from serologically positive Lyme disease patients that were negative at day 6 in culture were converted into long-term cultures. After 4 and 8 weeks spirochetal growth was identified in four of these ten cultures and those spirochetal structures stained positively with the polyclonal anti-Borrelia antibody. Examination of the slides showed individual spirochetes as well as small and large aggregates of spirochetes. Figure 4 shows representative fields of one collagen-coated slide from one of the positive samples (Panel A-D). To test whether a long-term culture can be set up directly, serum from seropositive Lyme disease patients was seeded into 45 ml of mBSK-H media in a Coplin jar containing two collagen slides. All attempts to establish a long-term Borrelia culture from patient sera in this manner were not successful (data not shown). This confirmed the need to seed a long-term culture from a short-term starter culture.
shows the immunocytochemical images of one representative clinical sample of a Lyme disease patient whose diagnosis was supported by the 2-tiered serological method that follows the CDC surveillance testing guidelines. The serum sample was cultured with our culture method for 8 weeks and the obtained collagen-coated slides were immunostained with polyclonal fluorescent antibodies against Borrelia burgdorferi. Panels A-D represent 4 different fields of one collagen-coated slide showing individual spirochetes and small and large spirochetal aggregates that stained positively for Borrelia burgdorferi antigen (green staining). Magnification: 200X.
Validation study in 72 CDC-positive Lyme disease patients
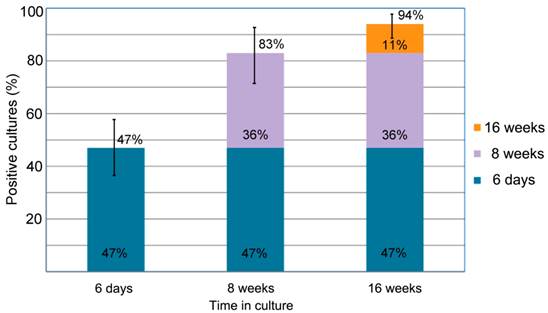
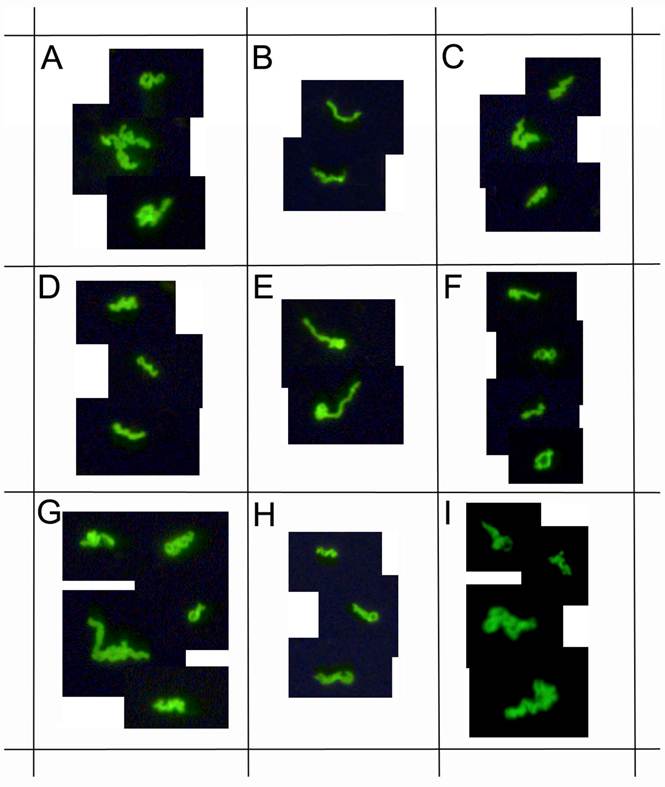
This study enrolled 72 Lyme disease patients whose diagnosis was confirmed by a typical clinical history and a positive result on a 2-tiered serological test following CDC testing guidelines. It was required that each donor had not been exposed to antibiotics for a minimum of 4 weeks prior to blood draw as recommended in previous studies [33] [34]. Short-term cultures were established in mBSK-H as described and after six days these cultures were used to seed collagen-supported long-term cultures. The details of the procedure are summarized in Figure 5. Analysis on day 6 using polyclonal anti-Borrelia antibody revealed that 34 of the 72 (47%) cultures showed growth (Figure 6). Long-term cultures were then begun from all the patient samples at day 6 and were analyzed again at 8 and 16 weeks (Figure 6). An additional 26 samples became positive for Borrelia at 8 weeks for a total of 60 of 72 (83%) and at 16 weeks an additional 8 samples became positive for Borrelia 68 of 72 (94%). All samples that were positive by polyclonal staining (representative images in Figure 7) also stained positively with the monoclonal antibody (representative images in Figure 8).
outlines the individual steps of the short and long-term culture procedures. ICC=immunocytochemistry.
summarizes the percentage of the successful Borrelia burgdorferi cultures of the clinical samples from the 72 Lyme disease patients whose diagnosis was supported by the 2-tiered serological method that follows the CDC surveillance testing guidelines. The samples were cultured with our novel method for 6 days, 8 weeks and 16 weeks and the cultures were confirmed to be Borrelia burgdorferi species by immunocytochemical and PCR methodologies (see Material and Methods). The figure shows that 34 of the 72 cultures were positive after 6-days of culturing (47% with a 95% confidence interval of 36%-58%) an additional 26 samples showed spirochetal presence at 8 weeks of culture (83% with a 95% confidence interval of 72%-92%). At 16 weeks an additional 8 samples became positive for spirochetal growth (94% with a 95% confidence interval of 86%-97%). The confidence interval values are depicted by bars.
demonstrates immunocytochemical images of nine representative positive clinical samples of the 72 Lyme disease patients whose diagnosis was supported by the 2-tiered serological method that follows the CDC surveillance testing guidelines. The samples were cultured with our novel method for 8 weeks and immunostained with polyclonal fluorescent antibodies against Borrelia burgdorferi (green staining). Panels A-I: Nine representative samples, 400x.

demonstrates immunocytochemical images of nine representative clinical samples of 72 Lyme disease patients whose diagnosis was supported by the 2-tiered serological method that follows the CDC surveillance testing guidelines. The samples were cultured with our novel method for 8 weeks and immunostained with monoclonal fluorescent antibodies against Borrelia burgdorferi (green staining). Panels A-I: Nine representative samples, 400x.
Borrelia-specific PCR products were amplified from each of the antibody-positive samples confirming that each sample contained Borrelia. Sequence analysis at the 16S rRNA or CTP synthase locus or both was performed and confirmed that each PCR product was Borrelia DNA. Significant sequence variations were identified among 51 samples sequenced at the CTP synthase gene locus indicating that samples were derived from independent sources and not from laboratory contamination. The percentage of the sequence identity of the clinical isolates to the Borrelia burgdorferi B31/N40 strains reference sequences (accession #AE000783 and #CP002228 respectively) ranged from 92%-100%. The numbers of polymorphic sites found in the clinical isolates for CTP synthase loci and the percentage of identity to Borrelia burgdorferi B31 sequence summarized in Table 3.
Summary of the numbers of the polymorphic sites for the CTP synthase locus and the % of identity to Borrelia burgdorferi B31 strain of the 51 clinical isolates obtained from our validation study.
| Number of Isolates | Genbank Accession Numbers of the Clinical Isolates | # of Polymorphic Sites | % of Identity to Borrelia burgdorferi B31 |
|---|---|---|---|
| 10 | JX867374, JX867377, JX867381 - 92, JX867395 - 97, JX867399, JX867400 and JX867405 | 0 | 100% |
| 1 | JX867419 | 2 | 99% |
| 1 | JX867376 | 21 | 97% |
| 1 | JX867393 | 39 | 93% |
| 21 | JX867375, JX867401 - 04, JX867406 - 16, JX867418 and JX867420 - 23 | 41 | 93% |
| 4 | JX867378 - JX867380 and JX867417 | 42 | 93% |
| 1 | JX867394 | 43 | 93% |
| 1 | JX867398 | 49 | 92% |
| 1 | JX867424 | 51 | 92% |
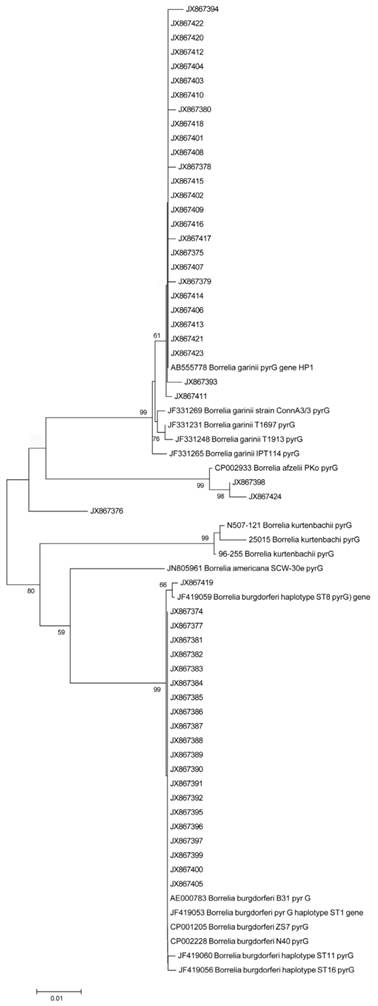
Sequence alignment (CLUSTELW) and neighbor-joining phylogenetic analysis were conducted on a 600 bp alignment of the CTP synthase gene of Bb clinical isolates obtained from the validation study using MEGA 5 Evolutionary Genetics Analysis program [36]. Tree support was evaluated by bootstrapping with 500 replications. Figure 9 shows the result of the analyses indicating of several sequence clusters for the CTP synthase genes derived from the DNA samples of the 51 clinical isolates. The resulting CTP synthase sequences were submitted to the NCBI national genome database (accession numbers are listed in Materials and Methods). Limited sequence variation was also noted at the well-conserved 16S rDNA site (data not shown).
shows a neighbor-joining phylogenetic analysis of 600 bp alignment of the CTP synthase gene (pyrG) of the 51 Borrelia clinical isolates obtained from our validation study using MEGA 5 Evolutionary Genetics Analysis program [36]. Alphanumeric codes represent GenBank (NCBI) ID numbers. Nucleotide sequences for CTP synthase locus for 51 clinical isolates have the following sequential accession numbers: JX867374 through JX867424. Numbers at nodes indicate bootstrap support values (500 replicates).
To ensure the specificity of this culture method and any subsequent detection system, serum was collected from 48 healthy controls with no history of tick bites or of Lyme disease and was processed using the above short-term and long-term culture methods. All 48 negative control samples were negative by dark field microscopy and by anti-Borrelia monoclonal and polyclonal antibody staining at all time points we examined (6 days, 8 weeks, and 16 weeks; 100%).
Discussion
Here we report the development and evaluation of an improved method to culture Borrelia spirochetes from peripheral blood of confirmed Lyme disease patients. Results from this study showed that by optimizing collection and transport conditions, media and growth environment, and the development of a long-term culture method together provided improved growth conditions for Borrelia. This is demonstrated by a 94% detection rate for Borrelia burgdorferi from sera of Lyme disease patients whose diagnoses were confirmed under the CDC surveillance testing guidelines. We have also found significant sequence variation in these Borrelia samples which further documents that the Borrelia grown in this culture system were not derived from laboratory contamination. The negative results from sera from healthy controls also argue against laboratory contamination as being the source of the positive cultures and further confirm the specificity of the test.
The maximum success rate for Borrelia blood cultures as outlined in recent publications is in the 40-44% range after 8-12 weeks of culturing [10] [30] [50]. By refining our collection and short-term cultivation methods we were able to get similar results after just six days in culture. The highest rate of Borrelia growth previously reported from clinical samples (up to 88%) involved cultures of skin, a tissue abundant in collagen [1] [2]. Such studies revealed a maximal success rate similar to what we achieved in our long term, collagen supported in vitro culture system (84% at 8 weeks and 94% at 16 weeks).
Unlike what has been reported by others, [50] we found poor results when blood specimens were collected in tubes containing EDTA. We found that the addition of BSK-H medium to the whole blood sample at the time of collection increased spirochete yield. As previously reported [44], we also noted that allowing the serum to separate, especially when mixed with BSK-H media, for up to 24 hours from the time of the blood draw improved the culture success rate. The BSK-H medium appeared to stabilize and even draw out the spirochetes from the cellular fraction. Presence of N-acetyl glucosamine (NAG) in the BSK-H medium, a known chemoattractant for Borrelia [51], may facilitate this separation and support the spirochetes during transport. Presumably, these freely suspended Borrelia were better able to be cultivated. Interestingly, there was a subset of clinical samples that grew better in the collection tubes without additional BSK media. Because of this observation, we utilized both methods in our experiments to ensure maximal yield.
BSK-H has been the preferred media for isolating and cultivating spirochetes from human clinical samples [1] [27]. We made four changes that include: 1. Higher concentration of rabbit serum (12% instead of 6%) incorporating the observation that cultivation of other Borrelia strains such as Borrelia hermsii requires a higher percentage of serum [52]. 2. Based on the previous observation that DTT added to BSK-H media helped to isolate Borrelia spirochetes from Ixodes ticks [37], by testing different concentrations of DTT we determined that a high concentration of DTT (100 μg/ml) enhanced the growth of Borrelia in our cultures. 3. Since Borrelia species are known to be microaerophilic, meaning that while some oxygen is needed for viability, high oxygen concentrations can be inhibitory [53], conditions that limit but not eliminate oxygen, such as partially shut lids, nearly full tubes and use of a CO2 incubator facilitated growth. 4. Low concentration of rifampicin (0.4-0.5 µg/ml) prevented secondary bacterial overgrowth in all but 2 out of 122 cultures used in this study. In addition, the use of two different types and sizes of tubes in the starter culture improved outcome.
The long-term culture system provided the greatest advantage in achieving a higher success rate. It was found that modified BSK-H media did not provide an efficient in vitro culture environment after 8-10 days. We thus investigated whether matrix protein as a solid support may provide a desirable environment for growth of Borrelia. [48]-[49]. Several matrix proteins were tested such as fibronectin, laminin and hyaluronan, but collagen gave the best results, consistent with previously published data [38] [54]. Interestingly, collagen is present both in skin biopsies and in the novel long-term culture presented here and may be a common reason for their growth advantage over other cultures.
The identity of the spirochetes was confirmed in all of the 68 culture-positive, CDC-positive patients enrolled in the validation study by staining positively with Bb-specific monoclonal and polyclonal immunostains. Additionally, further confirmation of the presence of Borrelia burgdorferi in these samples was accomplished by testing all clinical isolates with DNA PCR and then directly sequencing the DNA products. Again, in all cases, results were consistent with Borrelia burgdorferi.
Several measures were employed to rule out the possibility of false positives. Blood from 48 negative controls was cultivated using the above established methods and none showed any growth. Meticulous DNA analyses of the positive cultures including the use of both negative and positive controls in each PCR reaction did not reveal any evidence for contamination. To confirm that Borrelia isolates that were cultured from the 72 CDC positive patients were unique and to further prove that the PCR products were not from an environmental contaminant, PCR products were directly sequenced at either the 16S rRNA or the CTP synthase locus or both. The results confirmed that the sequences from each positive culture sample were derived from Borrelia. Sequence analysis within the amplified region of 16S rRNA gene showed only limited variation that is likely due to the highly conserved nature of 16S rRNA gene [55]. However, abundant sequence variations were noted at the CTP synthase locus from the positive cultures, indicating that the clinical isolates are unique and derived from wild-type Borrelia and not from a laboratory contaminant. The PCR and sequencing results were included in this study to further validate the polyclonal and monoclonal antibody assays used for the development of this Borrelia culture.
In summary, this report provides evidence for the value of a novel method for culturing Borrelia from human serum samples. The inclusion of key components such as modified culture media plus a unique culture environment has resulted in an improvement in the ability to cultivate this organism. This new culture method directly addresses the issue of the low numbers of Borrelia in clinical samples by amplifying their quantity through long term culture in which Borrelia were able to thrive for as long as eight months (data not shown). The versatility of this method allows for samples to be harvested from the culture at any point in time for further study, and it also serves as a source of Borrelia for a variety of direct detection techniques as well as for additional research. Finally, this unique culture method could play an important role in providing useful diagnostic information for select Lyme disease patients who might have tested negatively by other methods.
Acknowledgements
The authors thank Dr. Joseph Burrascano, Jr., Dr. Alan MacDonald and Dr. Parkash Gill for helpful discussion.
Competing Interests
This work was supported by a grant from RAM Capital LLC to the Research Division of Advanced Laboratory Services Inc (ALSI). During this study NP, AD, EAM, BAK were employees of ALSI and ES was a consultant. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors have declared that no competing interests exist.
References
1. Aguero-Rosenfeld ME, Wang G, Wormser GP. et al. Diagnosis of lymeborreliosis. Clin Microbiol Rev. 2005;18:484-509
2. Marques AR. Lyme Disease: A Review. Curr Allergy Asthma Rep. 2010;10:13-20
3. Bacon RM, Kugeler KJ, Mead PS. Surveillance for Lyme disease-United States, 1992-2006. MMWR Surveill Summ. 2008;57:1-9
4. Stanek G, Wormser GP, Strle F. et al. Lyme borreliosis. Lancet. 2012;379:461-73
5. Worsmer GP, Dattwyler RJ, Steere AC. et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43:1089-134
6. Liegner KB, Shapiro JR, Hogrefe W. et al. Recurrent erythema migrans despite extended antibiotic treatment with minocycline in a patient with persisting Borrelia burgdorferi infection. J American Acad Dermatol. 1993;28:312-314
7. Klempner MS, Hu LT, Johnson GM. et al. Two controlled trials of antibiotic treatment in patients with persistent symptoms and a history of Lyme disease. N Engl J Med. 2001;345:85-92
8. Dumler S. Molecular diagnosis of Lyme disease: review and meta-analysis. Mol Diagn. 2001;6:1-11
9. Nelson R. Controversy brewing over Lyme disease testing. Lancet InfectDis. 2005;5:605
10. Nowakowski J, Schwartz I, Aguero-Rosenfeld ME. et al. Laboratory diagnostic techniques for patients with early Lyme disease associated with erythema migrans: a composition of different techniques. Clin Infect Dis. 2001;33:2023-2027
11. Bunikis J, Barbour AJ. Laboratory testing for suspected Lyme disease. Med Clin North Am. 2002;86:311-340
12. Steere AC, McHugh G, Sikand VK. et al. Prospective study of serologic tests for lyme disease. Clin Infect Dis. 2008;47:188-195
13. Mejri N, Rutti B, Brossard M. Immunosuppressive effects of ixodes ricinus tick saliva or salivary gland extracts on innate and acquired immune response of BALB/c mice. Parasitol Res. 2002;88:192-197
14. Schuijt TJ, Coumou J, Fikrig E. et al. A tick mannose-binding lectin inhibits the vertebrate complement cascade to enhance transmission of the Lyme disease agent. Cell Host Microbe. 2011;10:136-146
15. Hastey CJ, Elsner RA, Baumgarth N. et al. Delays and diversions mark the development of B cell responses to Borrelia burgdorferi infection. J Immun. 2012;188:5612-5622
16. Hardin JA, Steere AC, Malawista SE. Immune complexes and the evolution of Lyme arthritis. Dissemination and localization of abnormal C1q binding activity. N Engl J Med. 1979;301:1358-1363
17. Coyle PK, Schutzer SE, Golightly MG. et al. Cerebrospinal fluid immune complexes in patients exposed to Borrelia burgdorferi: detection of Borrelia-specific and -nonspecific complexes. Ann Neurol. 1990;28:739-744
18. Schutzer SE, Coyle PK, Drulle J. et al. Sequestration of antibody to Borrelia burgdorferi in immune complexes in seronegative Lyme disease. Lancet. 1990;335:312-315
19. Zhong W, Oschmann P, Wellensiek HJ. Detection and preliminary characterization of circulating immune complexes in patients with Lyme disease. Med Microbiol Immunol (Berlin). 1997;186:153-158
20. Wallach FR, Forni AL, Steinberg CR. et al. Circulating Borrelia burgdorferi in patients with acute Lyme disease: results of blood cultures and serum DNA analysis. J Infect Dis. 1993;168:1541-1543
21. Nocton JJ, Bloom BJ, Rutledge BJ. et al. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in cerebrospinal fluid in Lyme neuroborreliosis. J Infect Dis. 1996;174:623-627
22. Rauter C, Mueller M, Hassler D. et al. Critical evaluation of urine-based PCR assay for diagnosis of Lyme borreliosis. Clin Diagn Lab Immunol. 2005;12:910-917
23. Klempner MS, Schmid CH, Hu LT. et al. Intralaboratory reliability of serologic and urine testing for Lyme disease. Am J Med. 2001;110:217-219
24. Al-Soud WA, Radstrom P. Purification and characterization of PCR inhibitory components in blood cells. J Clin Microbio. 2001;39:485-493
25. Molloy PJ, Persing DH, Berardi VP. False-positive results of PCR testing for Lyme disease. Clin Infect Dis. 2001;33:412-413
26. Aguero-Rosenfeld ME, Nadelman RB, Wormser GP. et al. Evolution of the serologic response to Borrelia burgdorferi in treated patients with culture-confirmed erythema migrans. J Clin Microbiol. 1996;34:1-9
27. Barbour AG. Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med. 1984;57:521-525
28. Liveris D, Wang G, Nowakowski J. et al. Quantitative detection of Borrelia burgdorferi in 2-millimeter skin samples of erythema migrans lesions: correlation of results with clinical and laboratory findings. J Clin Microbiol. 2002;40:1249-1253
29. Cerar T, Rusic-Sabljic E, Strle F. et al. Comparison of PCR methods and culture for the detection of Borrelia spp. in patients with erythema migrans. Clin Microbiol Infect. 2008;14:653-658
30. Liveris D, Nowakowski J, Wormser GP. et al. Comparison of five diagnostic modalities for direct detection of Borrelia burgdorferi in patients with early Lyme disease. Diagn Microbiol and Infect Dis. 2012;73:243-245
31. Casjens S, Palmer N, Stevenson B. et al. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 2000;35:490-516
32. Pollack R J, Telford III SR, Spielman A. Standardization of medium for culturing Lyme disease spirochetes. J Clin Microbiol. 1993;31:1251-1255
33. Nadelman RB, Nowakowski J, Wormser GP. et al. Failure to isolate Borrelia burgdorferi after antimicrobial therapy in culture-documented Lyme borreliosis associated with erythema migrans: report of a prospective study. Am J Med. 1993;94:583-588
34. Muellegger RR, Zoechling N, Kerl H. et al. No detection of Borrelia burgdorferi-specific DNA in erythema migrans lesions after minocycline treatment. Arch Dermatol. 1995;131:678-682
35. Margos G, Gatewood AG, Terekhova D. et al. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. PNAS. 2008;105:8730-8735
36. Tamura K, Peterson D, Nei M. et al. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28:2731-2739
37. Nelson JA, Bouseman JK, Bankowski MJ. et al. Isolation and characterization of Borrelia burgdorferi from Illinois Ixodes dammini. J Clin Microbiol. 1991;29:1732-1734
38. Sapi E, Luecke DF, Kaur N. et al. Evaluation of in vitro antibiotic susceptibility of different morphological forms of Borrelia burgdorferi. Infect Drug Resist. 2011;4:97-113
39. Brorson Ø, Brorson SH. In vitro conversion of Borrelia burgdorferi to cystic forms in spinal fluid, and transformation to mobile spirochetes by incubation in BSK-H medium. Infect. 1998;26:44-50
40. Gruntar I, Malovrh T, Cinco M. et al. Conversion of Borrelia garinii cystic forms to motile spirochetes in vivo. APMIS. 2001;109:383-388
41. Miklossy J, Kasas S, Yu S. et al. Persisting atypical and cystic forms of Borrelia burgdorferi and local inflammation in Lyme neuroborreliosis. J Neuroinflammation. 2008;5:40-58
42. Preac-Mursic V, Wanner G, Busch V. et al. Formation and cultivation of Borrelia burgdorferi spheroplast L-form variants. Infect. 1996;24:218-225
43. Preac-Mursic V, Wilske B, Reinhardt S. Culture of Borrelia burgdorferi on six solid media. Eur J Clin Microbiol Infect Dis. 1991;10:1076-1079
44. Phillips SE, Mattman LH, Moayad H. et al. A proposal for the reliable culture of Borrelia burgdorferi from patients with chronic Lyme disease, even from those previously aggressively treated. Infect. 1998;6:364-367
45. Rodríguez I, Lienhard R, Jouda F. et al. Evaluation of a modified culture medium for Borrelia burgdorferi sensu lato. Mem Inst Oswaldo Cruz Rio de Janeiro. 2007;102:999-1002
46. De Martino SJ, Sordet C, Piémont Y. et al. Enhanced culture of Borrelia garinii and Borrelia afzelii strains on a solid BSK-based medium in anaerobic conditions. Res Microbiol. 2006;157:726-729
47. Steere AC, Levin RE, Abraham JH 3rd. et al. Treatment of Lyme arthritis. Arthritis & Rheumatism. 1994;37:878-888
48. Zambrano MC, Beklemisheva AA, Cabello FC. et al. Borrelia burgdorferi binds to, invades and colonizes native type I collagen lattices. Infect Immun. 2004;72:3138-3146
49. Cabello FC, Godfrey HP, Newman SA. Hidden in plain sight: Borrelia burgdorferi and the extracellular matrix. Trends Microbiol. 2007;15:350-354
50. Wormser GP, Bittker S, Nadelman RB. et al. Comparison of the yields of blood cultures using serum or plasma from patients with early Lyme disease. J. Clin Microbiol. 2000;38:1648-1650
51. Bakker RD, Li C, Charon NW. et al. Identification of specific chemoattractants and genetic complementation of a Borrelia burgdorferi chemotaxis mutant: flow cytometry-based capillary tube chemotaxis assay. Appl Environ Microbio. 2007;73:1180-1188
52. Battisti JM, Raffel SJ, Schwan TG. A system for sites specific genetic manipulation of the relapsing fever spirochete Borrelia hermsii. Methods Mol Biol. 2008;431:69-84
53. Boylan JA, Lawrence KA, Gherardini FC. et al. Borrelia burgdorferi membranes are the primary targets of reactive oxygen species. Mol Microbiol. 2008;68:786-799
54. Sapi E, Bastian SL, Leucke DF. et al. Characterization of biofilm formation by Borrelia burgdorferi in vitro. PLoS ONE. 2012;7(10):e48277
55. Weisburg WG, Barn SM, Lane DJ. et al. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697-703
Author contact
![]() Corresponding author: : Eva Sapi Ph.D. Department of Biology and Environmental Science, University of New Haven, 300 Boston Post Road, West Haven CT 06510. Ph: 203-479-4552 Fax: 203-931-6097 Email: esapiedu.
Corresponding author: : Eva Sapi Ph.D. Department of Biology and Environmental Science, University of New Haven, 300 Boston Post Road, West Haven CT 06510. Ph: 203-479-4552 Fax: 203-931-6097 Email: esapiedu.