Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
5. Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2012; 9(10):923-932. doi:10.7150/ijms.4878 This issue Cite
Research Paper
Ischemic Postconditioning Protects Neuronal Death Caused by Cerebral Ischemia and Reperfusion via Attenuating Protein Aggregation
Jianmin Liang1, Jihang Yao2, Guangming Wang3, Ying Wang4, Boyu Wang3, Pengfei Ge3 ![]()
1. Department of Pediatrics, First Bethune Hospital of Jilin University, Changchun 130021, Jilin Province, P.R.China.
2. Grade 2010, Norman Bethune Medical School, Jilin University, Changchun 130021, Jilin Province, P.R.China.
3. Department of Neurosurgery, First Bethune Hospital of Jilin University, Changchun 130021, Jilin Province, P.R.China.
4. Department of Neurology, Second Affiliated Hospital of Kunming Medical Colledge, Kunming 650101, Yunnan Province, P.R.China.
Received 2012-7-15; Accepted 2012-11-8; Published 2012-11-23
Abstract
Objective: To investigate the effect of ischemic postconditioning on protein aggregation caused by transient ischemia and reperfusion and to clarify its underlying mechanism.
Methods: Two-vessel-occluded transient global ischemia rat model was used. The rats in ischemic postconditioning group were subjected to three cycles of 30-s/30-s reperfusion/clamping after 15min of ischemia. Neuronal death in the CA1 region was observed by hematoxylin-eosin staining, and number of live neurons was assessed by cell counting under a light microscope. Succinyl-LLVY-AMC was used as substrate to assay proteasome activity in vitro. Protein carbonyl content was spectrophotometrically measured to analyze protein oxidization. Immunochemistry and laser scanning confocal microscopy were used to observe the distribution of ubiquitin in the CA1 neurons. Western blotting was used to analyze the quantitative alterations of protein aggregates, proteasome, hsp70 and hsp40 in cellular fractions under different ischemic conditions.
Results: Histological examination showed that the percentage of live neurons in the CA1 region was elevated from 5.21%±1.21% to 55.32%±5.34% after administration of ischemic postconditioning (P=0.0087). Western blotting analysis showed that the protein aggregates in the ischemia group was 32.12±4.87, 41.86±4.71 and 34.51±5.18 times higher than that in the sham group at reperfusion 12h, 24h and 48h, respectively. However, protein aggregates were alleviated significantly by ischemic postconditioning to 2.84±0.97, 13.72±2.13 and 14.37±2.42 times at each indicated time point (P=0.000032, 0.0000051 and 0.0000082). Laser scanning confocal images showed ubiquitin labeled protein aggregates could not be discerned in the ischemic postconditioning group. Further study showed that ischemic postconditioning suppressed the production of carbonyl derivatives, elevated proteasome activity that was damaged by ischemia and reperfusion, increased the expression of chaperone hsp70, and maintained the quantity of chaperone hsp40.
Conclusion: Ischemic postconditioning could rescue significantly neuronal death in the CA1 region caused by transient ischemia and reperfusion, which is closely associated with suppressing the formation of protein aggregation.
Keywords: Ischemic postconditioning, Protein aggregation, Proteasome, Chaperone
1. Introduction
Transient global cerebral ischemia is a clinical outcome related to cardiac arrest, cardiopulmonary bypass surgery and other situations that deprives the oxygen and glucose in brain during a short period [1, 2]. In the rat model simulating human transient global cerebral ischemia, it was found that neurons in the hippocampus CA1 region were severely damaged during the period of reperfusion, which leaded to the impairment of rats' memory or spatial learning [3]. Similarly, in human being, a clinical study showed that the lesion in CA1 region could result in place learning deficit [4]. These indicate that protection of neuronal injury or death in the CA1 region would improve the prognosis of the patients with transient ischemia and reperfusion.
Protein aggregation is a biological phenomenon in which abnormal proteins accumulate and clump together either intra- or extracellularly [5]. Until Hu et al found that abnormal proteins accumulated in the neurons destined to die after transient cerebral ischemia [6], protein aggregation had been thought to play a pivotal role in neurodegenerative disease such as Parkinson's disease and Alzheimer's disease [7]. Currently, as well as other mechanisms such as calcium overload and oxidative stress, protein aggregation is also regarded as one of the important pathological features of cerebral ischemia and reperfusion [8]. Although these aggregated proteins cannot function normally, they could stick to the intracellular membrane structures and result in cell losing its normal function and dying in the end [6]. Therefore, aggregated proteins should be rescued or eliminated in order to keep neurons alive and function normally.
Recently, it had been reported that ischemic postconditioning has protective effects on neuronal injury or death caused by focal or global ischemia and reperfusion [9, 10]. Ischemic postconditioning is defined as a series of rapid intermittent interruptions of blood flow in the early phase of reperfusion that mechanically alters the hydrodynamics of reperfusion [9]. It could be performed before re-establishment of blood supply to brain, which makes it become a feasible method used potentially in clinical practice. Despite studies showed that ischemic postconditioning could rescue neuronal injury or death via inhibiting inflammation, suppressing oxidative stress or preventing apoptosis [11, 12], the effects of ischemic postconditioning on protein aggregation are still unclear. Studying the alteration of protein aggregation would contribute further to clarify the protective mechanism of ischemic postconditioning. Therefore, in the current study, we investigated the role of ischemic postconditioning in protein aggregation by using rat model of transient global ischemia.
2. Materials and methods
2.1 Materials
Anti-hsp70 and anti-hsp40 polycolonal antibody were from Cell Signaling Company (Beverly, MA, USA). Anti-ubiquitin monoclonal antibody was from Chemicon Company (Temecula, CA, USA). Anti-20S-α-1 and anti-19S S12 were from Affiniti Research Products Company (Plymouth Meeting, Pa, USA). Propidium iodide and fluorescence-labeled anti-mouse and anti-rabbit IgG were from Jackson Immunoresearch Laboratory (West Grove, PA, USA). Protein concentration assay kit was from Bio-rad Laboratory (Hercules, CA, USA). ECL Western blot detection reagents were from Amersham Company (Piscataway, NJ, USA). Actin-β and other reagents were from Sigma Company (St. Louis, MO, USA). PVDF membranes were from Millipore Company (Billerica, MA, USA). Light microscope was Olympus BX-51, laser scanning confocal microscope was Zeiss LSM 510, and vebrotome was Leica VT-1000S.
2.2 Animals
Adult male Wistar rats (weighing 280-300g) supplied by Jilin University Experimental Animal Center were housed in a temperature-controlled room (22-25◦C) with free access to food and water on a 12-h light/dark cycle. All animal procedures were approved by the ethical committee for animal experiments, Jilin University, Changchun, China.
2.3 Ischemia model
Brain ischemia was produced by using the 2-vessel occlusion (2VO) model. Male Wistar rats were fasted overnight and freely accessed to water. Anesthesia was induced with 5% halothane and maintained with 2% halothane in oxygen/nitrous oxide (30%/70%) gas mixture. Catheters were inserted into the femoral artery and tail artery to allow blood sampling, arterial blood pressure recording and drug infusion. Both of the common carotid arteries were encircled by loose ligatures. At 10 min prior to ischemia induction and 10 min post-ischemia, blood gases were measured and adjusted to PaO2 > 90 mmHg, PaCO2 35-45 mmHg, and pH 7.35-7.45 by regulating the tidal volume of the respirator. Bipolar electroencephalograph was recorded before, during and after ischemia until rat recovery from anesthesia. Brain ischemia was induced by withdrawing blood via the femoral artery catheter to produce a mean artery blood pressure of 50 mmHg, followed by clamping both carotid arteries for 15 min. Mean artery blood pressure was maintained at 50 mmHg during the ischemic period by withdrawing or infusing blood through the femoral catheter. At the end of the ischemia, the clamps were removed and the withdrawn blood was re-infused. Halothane was discontinued and all wounds were sutured. In all experiments, brain temperature was maintained at 37 oC before, during and after ischemia (15 min of reperfusion).
2.4 Postconditioning protocol
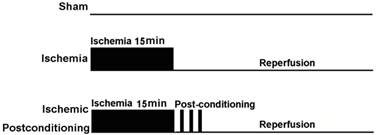
At the start of the study, 150 rats were assigned randomly into an ischaemia group and an ischemic postconditioning group according to a computer generated randomization schedule. On the basis of reperfusion time, each group was divided into following subgroups, including sham-operated group (as control), reperfusion 12h group, reperfusion 24h group, reperfusion 48h group and reperfusion 72h group. Each group included four rats at least. Except the rats in the sham-operated group did not subject to clamp both carotid arteries, other rats were subjected to cerebral ischaemia as described above. Additionally, the rats in ischemic postconditioning group were subjected to three cycles of 30-s/30-s reperfusion/clamping after 15min of ischaemia (Fig. 1).
Schematic depicting the order of surgical procedures for Wistar rats undergoing sham operation, transient ischemia and ischaemia plus ischemic postconditioning (ischemic postconditioning consisted of 3 cycles of 30-s/30-s reperfusion/ clamping after 15min ischemic episode).
2.5 Brain tissue fixation
After rat was anesthetized, the thorax was opened and the heart was disclosed. Herpin (0.1 mL, 300 IU/kg) was injected into the left ventricle before the catheter was inserted into the main artery via left atrium. Then, PBS was perfused into the vascular system at 4 oC for 3 min, and PBS with 4% paraformaldehyde was perfused at 4 oC for another 3 min. Subsequently, the brain tissue was taken out and put into PBS fixation solution containing 4% paraformaldehyde at 4 oC. Twelve hours later, the 13-µm and 50-µm coronal brain slices were cut by vibrotome and the brain slices in similarity were selected for hematoxylin-eosin (HE) staining and immunohistochemistry labeling, respectively.
2.6 Hematoxylin and Eosin staining and histological examination
The 13-µm brain slices were mounted on slides and kept in dark room until dried. These dried slices were immersed in distilled water for 1 min prior to dehydration in gradient ethanol solution. Then, they were put into hematoxylin solution for 15 s, washed up by distilled water, immersed in Scott solution for 10 s, and washed again by distilled water. After being immersed in Eosin solution 10 s, these brain slices were redehydrated in gradient ethanol solution again, treated with dimethylbenzene and covered with coverslips. Under 40× objective lens of light microscope, the living neurons were counted from the representative sections of hippocampus CA1 region by an observer blinded to the treatment. Total number of viable neurons per mm was considered for expressing hippocampal CA1 neuronal density.
2.7 Immunohistochemical analysis
The 50-µm brain slices were put into 24 wells plate with 500 µL citrate buffer (pH 6.0) and incubated in a 100 oC water bath for 10 min. After cooling down, the slices were washed with the TBS solution containing 0.2% TX-100 for 10 min and blocked in the TBS solution containing 3% BSA and 0.2% TX-100 for 1 h at room temperature. Then, they were incubated with 1:1000 monocolonal anti-ubiquitin antibody at 4 oC overnight. After washed up with TBS solution containing 0.1%TX-100 for three times (10 min each) at room temperature, the antibody-labeled brain slices were incubated with 1% BSA containing fluorescence labeled anti-mouse IgG (1:200) and propidium iodide (1:1 000) for 1 h at room temperature. Again, the brain slices were washed with TBS solution containing 0.1% TX-100 for three times (10 min), and then they were mounted on slides and dried in dark room. Mounting medium was added on the slides prior to be covered with coverslips for observing by a laser scanning confocal microscope.
2.8 Cytosolic fractions and protein aggregates isolation
As describe previously [13], after rats were anesthetized, the heads were cut and the brain tissues were taken out on dry ice. After the brain tissue was sliced, the CA1 region tissue were isolated under a microscope and frozen in liquid nitrogen and kept in -70oC refrigerator. After weighting, the tissues were homogenized with Dounce homogenizer in 10 volumes of ice-cold homogenization buffer (1.5 mmol/L Tris base-HCl pH 7.6, 1 mmol/L DTT, 0.25 mol/L sucrose, 1 mmol/L MgCl2, 1.25 µg/mL pepstatin A, 10 µg/mL leupeptin, 2.5 µg/mL aproptonin, 0.5 mmol/L PMSF, 2.5 mmol/L EDTA, 1 mmol/L EGTA, 0.1 mol/L Na3VO4, 50 mmol/L NaF, and 2 mmol/L sodium pyrophosphate). The homogenate was centrifuged at 800 g for 10 min at 4 oC to obtain the pellets and supernatants. The supernatants were further centrifuged at 10 000 g for 20 min at 4 oC to obtain the second supernatants and second pellets. The second supernatants were centrifuged at 165 000 g for 1 h at 4 oC to obtain the final supernatants (cytosolic fraction). The second pellet fractions were suspended in the homogenization buffer containing 1% TX100 and 400 mmol/L KCl, sonicated for 5 s for 3 times, washed on a shaker for 1 h at 4 oC, and then centrifuged at 20 000 g for 10 min at 4 oC to obtain TX -insoluble fractions (protein aggregates). After the protein concentrations were measured, the cytosolic fraction and protein aggregates were kept in -20 oC refrigerator.
2.9 Proteasome activity assay
Ten microliters (1 µg/µL) of each freshly made supernatant (S2) from CA1 tissue was incubated in a 96-well plate at 37°C for 30 minutes with 10 µL of 300 µmol/L of succinyl-LLVY-AMC substrate and 85 µL of assay buffer (20 mmol/L Tris-HCl, pH 7.5, and 20% glycerol). Release of fluorescent AMC was measured with a spectrofluorometer (Perkin-Elmer Life and Analytical Sciences, Inc, Wellesley, Mass) at 440 nm with an excitation wavelength of 380 nm. The absorbance ratio of each group to sham group was used to show the variation.
2.10 Assay of protein carbonyl levels
Protein carbonyl content was measured spectrophotometrically according to the procedure described by Jolitha et al [14]. Briefly, 10 mmol dinitro-phenyl-hydrazine in 2.5 M HCl was added to the tissue homogenate from CA1 region tissue and incubated in dark for 60 min at room temperature. This was followed by vortex mixing, addition of 20% trichloroacetic acid (w/v) and subsequently washing three times with ethanol/ethyl acetate (1:1 v/v) mixture. Precipitated proteins were then re-dissolved in 6 M guanidine HCl in 20 mmol phosphate buffer (pH 6.5). Insoluble substances were removed by centrifugation and absorbance of the supernatant was read at 370 nm. Protein contents of the supernatant fractions were determined by the Bradford assay. The absorbance ratio of each group to sham group was used to show the variation.
2.11 Western blot analysis
Western blot analysis was conducted with 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Protein samples for Western blot contained 50 µg of cytosolic fractions, or 20 µg of protein aggregates. Four samples (from four different rats) in every experimental group were used for statistical analysis. After electrophoresis, proteins were transferred onto PVDF membranes. The membranes were probed with anti-ubiquitin (1:2 000), anti-20Sα-1(1:1000), anti-19S S12(1:1000), anti-hsp40(1:500), or anti-hsp70 (1:1 000) antibody. The membranes were then incubated with anti-mouse (1:20 000) or anti-rabbit (1:50 000) secondary antibody for 1 h at room temperature. The blots were developed with ECL detection method, and the optical density of protein band was quantified using Kodak 1D gel analysis software. The value of the optical density in sham group was used as the baseline reference value, and the ratio of each group to sham group represented the changes.
2.12 Data analysis
All data are expressed as Mean ± SD and analyzed statistically by SPSS 17.0 software (SPSS Corp, USA). Student's t-test was used as appropriate for comparison between different groups. P < 0.05 was considered statistically significant.
3. Results
3.1 Ischemic postconditioning improved living neurons in CA1 region
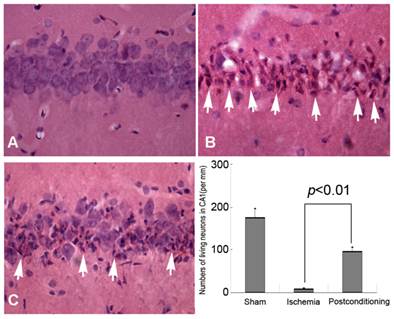
HE staining and Histological examination is an effective and easy way to evaluate cell viability, and it was used in this study to evaluate delayed neurons death after different ischemic conditions. Generally, compared with the normal neurons, the dead or dying neurons presented polygonal condensed nucleus and pink cytosl [15]. As shown in figure 2, only 9.12±1.87 neurons per mm in CA1 region were alive after 72h reperfusion in the ischemia group, whereas 96.85±9.71 neurons per mm alive in the ischemic post-conditioning group (P =0.0082 vs Sham group). Therefore, after 3 cycles of 30-second/30-second reperfusion/reclampping, many neurons in the CA1 region were rescued from subsequent lethal ischemia. This result demonstrated three cycles of 30 seconds reperfusion is an effective ischemic post conditioning procedure.
Morphological examinations of CA1 neurons by Hematoxylin and Eosin staining (40×). A, sham group; B, ischemia group; C, ischemic postconditioning group. The dead or dying neurons presented polygonal condensed nucleus and pink cytosol (arrow head). Statistics showed the living neurons in ischemic postconditioning group were 96.85±9.71 per mm, which was markedly higher than 9.12±1.87 per mm of the ischemia group. Each group contained four rats.
3.2 Ischemic postconditioning attenuated protein aggregation
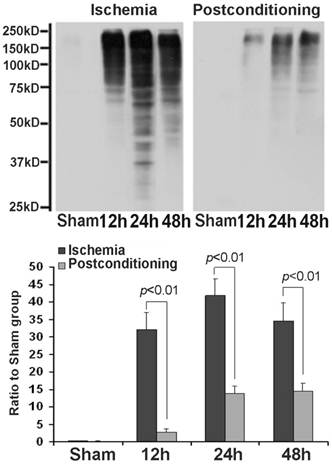
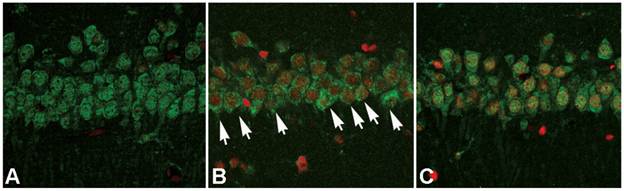
After cellular fractions were isolated, the TX-100 insoluble fraction was regarded as protein aggregates, and anti-ubiquitin antibody could be used to detect the formation of protein aggregates [6]. As figure 3 showed, at reperfusion 12h, 24h and 48h, the protein aggregates was 32.12±4.87, 41.86±4.71 and 34.51±5.18 times higher than that in the sham group. However, after administration of ischemic postconditioning, they reduced to 2.84±0.97, 13.72±2.13 and 14.37±2.42 times at each indicated time point (P=0.000032, 0.0000051 and 0.0000082 vs contemporary ischemia group). Because the quantity of protein aggregates culminated at reperfusion 24h (Fig 3) and part of CA1 neurons died at reperfusion 48h (data not shown), we chose to observe the distribution of protein aggregates at reperfusion 24h. Laser scanning confocal images of the ischemia group showed that proteins aggregates labeled by ubiquitin were located in cytosol and around nucleus, but they could not be discerned in the ischemic postconditioning group (Fig 4). These data suggested that the protein aggregation was attenuated by ischemic postconditioning performed before reperfusion.
The formation of protein aggregates detected by Western blotting. At reperfusion 12h, 24h and 48h, the protein aggregates was 32.12±4.87, 41.86±4.71 and 34.51±5.18 times higher than that in the sham group. However, after administration of ischemic postconditioning, they reduced to 2.84±0.97, 13.72±2.13 and 14.37±2.42 times at each indicated time point. The CA1 tissue used in each group were from 8 rats.
The distribution of protein aggregates at reperfusion 24h under laser scanning confocal microscope. The images of the ischemia group showed that proteins aggregates labeled by ubiquitin were located in cytosol and around nucleus (arrow), but they could not be discerned in the sham and ischemic postconditioning groups. A: sham group; B: ischemia group; C: ischemic postconditioning group. Each group contained 4 rats.
3.3 Ischemic postconditioning reduced the products of protein oxidation
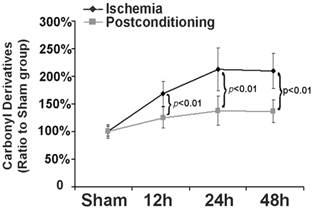
Part of the components of protein aggregates are from oxidized proteins [16], thus we examined in this study the effect of ischemic postconditioning on protein oxidation by measuring carbonyl derivatives of protein proline, lysine, arginine and threonine residues, a kind of common products of protein oxidation. As shown in figure 5, in comparison with that of the sham group, the levels of carbonyl derivatives increased to 169.72%±23.16%, 214.33%±39.41% and 210.56%±32.11% at reperfusion 12h, 24h and 48h, respectively. However, they decreased to 125.24%±19.72%, 139.68%±27.54%, and 137.18%±22.97 (P=0.0076, 0.00048 and 0.0037 versus corresponding group) after treatment with ischemic postconditioning. This indicated that inhibition of protein oxidation is one pathway via which ischemic postconditioning attenuated the formation of protein aggregates.
Measurement of level of carbonyl derivatives. Carbonyl derivatives in the ischemia group increased with elongation of reperfusion time, which was significantly suppressed at each scheduled time point by administration of ischemic postconditioning performed before reperfusion. The CA1 tissues used for analysis were from 8 rats in each group.
3.4 Ischemic postconditioning preserved proteasome activity
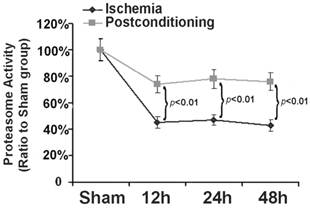
Within cells, ubiquitin proteasome system is a selective pathway to degrade intracellular abnormal proteins. Therefore, as described before [17], we also selected Succinyl-LLVY-AMC, a specific substrate of proteasome for assay in vitro, to measure the effects of ischemic postconditioning on proteasome activity. As shown in figure 6, in comparison with that in sham group, proteasome activity reduced to 45.32%±4.13%, 47.41%±3.83%, and 43.52%±4.22% at reperfusion 12 h, 24h and 48h following ischemia, respectively. Nevertheless, it maintained at 73.63%±6.61%, 79.23%±7.14%, and 77.63%±5.92% (P=0.0031, 0.0019 and 0.0013 versus corresponding groups) after application of three cycles of 30s ischemia and 30s reperfusion at the end of ischemia. Our results showed that ischemic postconditioning could partly reverse the reduction of proteasome activity induced by ischemia/reperfusion.
Assay of proteasome activity. Proteasome activity was damaged markedly from reperfusion 12h, but it was maintained at higher level by ischemic postconditioning. The CA1 tissues used for analysis were from 8 rats in each group.
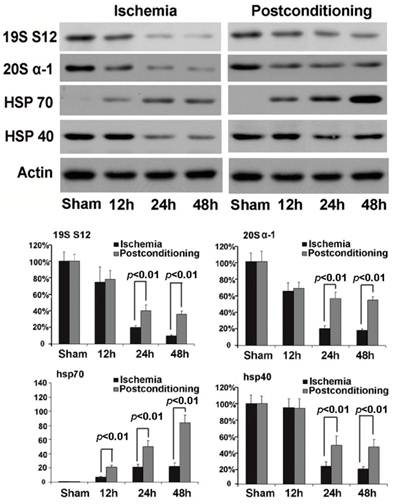
In order to find out the mechanism underlying the maintaining of proteasome activity after application of ischemic postconditioning, we examined the quantitative alteration of proteasome in the CA1 neurons by Western blotting (fig 7). We found that the quantity of 20S (the core unit of proteasome) and 19S (catalytic unit of proteasome) reduced at reperfusion 12h and continued to 48h in the ischemia group. By contrast, ischemic postconditioning attenuated the quantitative reduction of 20S and 19S (P=0.00062 and 0.00059 for 20S, and P=0.0092 and 0.0078 for 19S). This indicated that ischemic postconditioning maintained the quantity of proteasome in the CA1 neurons that suffered subsequently lethal ischemia, which might partly underlie why ischemic postconditioning could depress the reduction of proteasome activity.
Western blotting analysis of the quantitative alterations of proteasome, hsp70 and hsp40. In the ischemia group, 20S quantitatively decreased to 65.23%±9.65%, 20.37%±3.78%, and 18.15%±1.77%, and19S decreased to 74.89%±17.67%, 20.16±2.23%, and 11.32±1.21% at reperfusion 12h, 24h, and 48h, respectively. By contrast, ischemic postconditioning maintained 20S to 67.85%±7.16%, 56.27%±7.82%, and 53.78%±4.24%, and 19S to 78.34%±11.28%, 41.56%±7.09%, and 36.33%±3.98% at reperfusion 12h, 24h, and 48h. Despite chaperone hsp70 increased to 7.08±1.12, 20.79±4.29, and 22.91±4.85 times at reperfusion 12h, 24h, and 48h than that in the sham group, it was increased by ischemic postconditioning much more to 21±2.96, 50±8.26, and 105±11.18 times at each indicated time point. The hsp40 expression down-regulated to 22.38%±5.63% and 18.89%±2.83% at reperfusion 24h and 48h in the ischemia group, but ischemic postconditioning maintained its expression to 48.22%±11.77% and 45.98%±8.91% at reperfusion 24h and 48h. The CA1 tissues used for analysis were from 8 rats in each group.
3.5 ischemic postconditioning maintained the quantity of hsp40 and up-regulated the expression of hsp70
Chaperones are a functionally related group of proteins that assist protein folding under physiological and stress conditions [18, 19]. In this study, we found that the expression of chaperone hsp70 was up-regulated with elongation of reperfusion in ischemia group, but ischemic postconditioning made its expressional increment much more (P=0.0075, 0.0061 and 0.000032 at 12h, 24h, and 48h versus corresponding groups ). For chaperone hsp40, despite its expression was down-regulated at reperfusion 24h and 48h in both the ischemia group and the ischemic postconditioning group, its quantity in ischemic postconditioning group was significantly higher than that in the ischemia group (P=0.0059 and 0.0066 at 24h and 48h versus corresponding groups). Our results showed that ischemic postconditioning improved the content of hsp70 and hsp40 in the CA1 neurons, which might be a factor affecting the reduction of protein aggregation induced by ischemia and reperfusion.
4. Discussion
The protective effects of ischemic postconditioning on ischemic injury have been found in various organs, such as brain, liver and intestine [8, 9, 20]. Its underlying mechanism has also been investigated widely and was found to be associated with multiple factors involved in the pathological course of delayed neuronal death caused by ischemia and reperfusion, such as suppression of oxidative stress, activation of signal pathway, regulation of enzyme activity and inhibition of endoplasmic stress [21-26]. Despite previous studies had shown that protein aggregation is an important factor affecting neuronal injury and death caused by ischemia and reperfusion [6, 8], the effects of ischemic postconditioning on protein aggregation has not yet been clear. In this study, by using rat model of transient global ischemia, we demonstrated that the benefit of ischemic postconditioning is associated with attenuating protein aggregation.
Overproduction of abnormal proteins that exceeds the capacity of protein degradation is an important factor leading to the formation of cytotoxic protein aggregates. It is currently thought that unfolded proteins, misfolded proteins and oxidized proteins constitute the majority of abnormal proteins [27]. How abnormal proteins are generated under ischemia and reperfusion condition is still uncertain, but it was thought previously that ischemic stress made cellular proteins unfold and lose their three dimensional conformation, so that the sticky hydrophobic residues buried within the proteins are exposed and tend to stick to one another, precipitate, and accumulate [6, 28].
Similar to our study, protein oxidation under the condition of transient ischemia and reperfusion was previously studied by using rat model. The results showed that the carbonyl derivatives, a kind of products of protein oxidation, steadily increased in re-flow period and culminated at 48 h of reperfusion [29]. Moreover, it was found that oxidized proteins are prone to accumulating together and forming cytotoxic protein aggregates [30, 31]. We did not investigate in this study the mechanism that underlies the attenuation of protein oxidation after application of ischemic postconditioning, but our previous study showed that ischemic postconditioning limited the increment of oxidized protein through inhibiting oxidative stress [32]. Recently, it was found that protein aggregates did not form at the end of ischemia, but formed during the period of reperfusion [33], which made us speculate that ischemic postconditioning could disturb the formation course of protein aggregates. In this study, we proved that ischemic postconditioning suppressed protein aggregation and protein oxidation, indicating that ischemic postconditioning rescued neuronal death in the CA1 region via limiting the formation of protein aggregation.
Decreased capacity of intracellular protein degradation system is another factor influencing protein aggregation. There are two major protein degradation systems within cells, the proteasome pathway and the autophagy pathway [34]. Although the function of autophagy in the neuronal death caused by ischemia and reperfusion is still unclear, the ubiquitin immunoreactivity of protein aggregates suggested that proteasome is a crucial pathway for degrading these abnormal proteins prior to their aggregation [35]. Moreover, it was found that the decline of proteasome activity is not able to remove oxidized proteins efficiently from old cells and proteasomal inhibition caused the formation of protein aggregates [36, 37]. Because previous studies showed that proteasome activity could be damaged by transient ischemia and reperfusion [17, 38], we compared the proteasome activity in the CA1 neurons treated with or without ischemic postconditioning. Our results showed that, ischemic postconditioning kept proteasome activity at higher level via maintaining its quantity, which contributed to degrade more unfolded, misfolded and oxidized proteins. Thus, modulation of proteasome activity by ischemic postconditioning is another reason leading to the reduction of protein aggregation.
However, it was also reported that a proteasome inhibitor given to rats at the end of 2 h focal ischaemia would decrease the size of cerebral infarct [39]. Current studies suggest that excessive inhibition of proteasome activity might activate the anti-inflammatory effect linked to the modulation of nuclear factor activity, attenuate the expression of cytokine and cellular adhesion molecules, and reduce the infiltration of neutrophil and macrophage into the injured rat brain [40]. Additionally, an in vitro study showed that inhibition of proteasome activity would result in activation of autophagy pathway, which could eliminate oxidized proteins and might explain why a proteasome inhibitor provides protection for cerebral injury induced by focal ischaemia and reperfusion [41]. By contrast, the study by Liu et al showed that the activity of autophagy might be suppressed under the condition of ischemia and reperfusion [42], which indicated that suppression of autophagy might be another mechanism underlying the formation of protein aggregates following ischemia and reperfusion. Especially, recent study demonstrated that autophagy played an important role in the cardio-protection induced by ischemic preconditioning [43], suggesting maintaining autophagy is a potential strategy to prevent protein aggregation. Thus, it should be investigated in future whether the reduction in protein aggregates caused by ischemic postconditioning is via autophagy.
The cellular machinery for maintaining proteostasis includes molecular chaperones as well, which facilitate protein folding and reduce proteotoxicity. In addition to their role in protein folding, chaperones help assemble and disassemble protein complexes, help present substrates for degradation, and suppress protein aggregation [44]. Among the chaperones, hsp70 is a stress-induced protein located in cytoplasm and has the ability to recognize and bind nascent, partially folded and unfolded proteins, and promote their refolding or degradation, while hsp40 could enhance this process [44]. Thus, in combination with hsp40, hsp70 could prevent protein aggregation and facilitate correct protein folding. In this study, our results suggested that suppression of protein aggregation by ischemic postconditioning might be related to elevate the expression of inducible chaperone hsp70 and maintained the quantity of hsp40.
5. Conclusions
Our study showed that ischemic postconditioning could reduce significantly neuronal death in the CA1 region caused by transient ischemia/reperfusion, which is closely associated with suppressing the formation of protein aggregation. The attenuation of protein aggregation is associated with decreasing the production of protein oxidization, maintaining the activity and quantity of proteasome, limiting the reduction of hsp40, and up-regulating the expression of hsp70. Our study indicated that ischemic postconditioning is an effective method to rescue the neuronal death caused by ischemia and reperfusion.
Acknowledgements
This work was supported by National Nature and Science Foundation (30973110) and (81171234), and the outstanding youth grant (20080139) from the science and technology department of Jilin Province.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Taraszewska A, Zelman IB, Ogonowska W, Chrzanowska H. The pattern of irreversible brain changes after cardiac arrest in humans. Folia Neuropathol. 2002;40:133-41
2. Tanabe M, Watanabe T, Ishibashi M, Hirano N, Tabuchi S, Takigawa H. Hippocampal ischemia in a patient who experienced transient global amnesia after undergoing cerebral angiography. Case illustration. J Neurosurg. 1999;91:347
3. Ruan YW, Han XJ, Shi ZS, Lei ZG, Xu ZC. Remodeling of synapses in the CA1 area of the hippocampus after transient global ischemia. Neuroscience. 2012;218:268-77
4. Bartsch T, Schönfeld R, Müller FJ, Alfke K, Leplow B, Aldenhoff J, Deuschl G, Koch JM. Focal lesions of human hippocampal CA1 neurons in transient global amnesia impair place memory. Science. 2010;328:1412-5
5. Aguzzi A, O'Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237-48
6. Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20:3191-9
7. Mengesdorf T, Jensen PH, Mies G, Aufenberg C, Paschen W. Down-regulation of parkin protein in transient focal cerebral ischemia: A link between stroke and degenerative disease? Proc Natl Acad Sci U S A. 2002;99:15042-7
8. Ge P, Luo Y, Wang H, Ling F. Anti-protein aggregation is a potential target for preventing delayed neuronal death after transient ischemia. Med Hypotheses. 2009;73:994-5
9. Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114-21
10. Wang JY, Shen J, Gao Q, Ye ZG, Yang SY, Liang HW, Bruce IC, Luo BY, Xia Q. Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke. 2008;39:983-890
11. Xing B, Chen H, Zhang M, Zhao D, Jiang R, Liu X, Zhang S. Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke. 2008;39:2362-2369
12. Xing B, Chen H, Zhang M, Zhao D, Jiang R, Liu X, Zhang S. Ischemic post-conditioning protects brain and reduces inflammation in a rat model of foal cerebral ischemia/reperfusion. J Neurochem. 2008;105:1737-1745
13. Liu CL, Ge PF, Zhang F, Hu BR. Co-translational protein aggregation after transient cerebral ischemia. Neuroscience. 2005;134:1273-84
14. Jolitha AB, Subramanyam MV, Asha Devi S. Modification by vitamin E and exercise of oxidative stress in regions of aging rat brain: studies on superoxide dismutase isoenzymes and protein oxidation status. Exp Gerontol. 2006;41:753-63
15. Ge PF, Luo TF, Zhang JZ, Chen DW, Luan YX, Fu SL. Ischemic preconditioning induces chaperone hsp70 expression and inhibits protein aggregation in the CA1 neurons of rats. Neurosci Bull. 2008;24:288-96
16. Norris EH, Giasson BI. Role of oxidative damage in protein aggregation associated with Parkinson's disease and related disorders. Antioxid Redox Signal. 2005;7:672-84
17. Ge P, Luo Y, Liu C, Hu B. Protein Aggregation and Proteasome Dysfunction after Brain Ischemia. Stroke. 2007;38:3230-6
18. Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571-9
19. Ohtsuka K, Hata M. Molecular chaperone function of mammalian Hsp70 and Hsp40 - a review. Int J Hyperthermia. 2000;16:231-45
20. Liu KX, Li YS, Huang WQ, Chen SQ, Wang ZX, Liu JX, Xia Z. Immediate postconditioning during reperfusion attenuates intestinal injury. Intensive Care Med. 2009;35:933-42
21. Zhang W, Wang B, Zhou S, Qiu Y. The effect of ischemic post-conditioning on hippocampal cell apoptosis following global brain ischemia in rats. J Clin Neurosci. 2012;19:570-3
22. Guo JY, Yang T, Sun XG, Zhou NY, Li FS, Long D, Lin T, Li PY, Feng L. Ischaemic postconditioning attenuates liver warm ischaemia-reperfusion injury through Akt-eNOS-NO-HIF pathway. J Biomed Sci. 2011;18:79
23. Zhou C, Tu J, Zhang Q, Lu D, Zhu Y, Zhang W, Yang F, Brann DW, Wang R. Delayed ischemic postconditioning protects hippocampal CA1 neurons by preserving mitochondrial integrity via Akt/GSK3β signaling. Neurochem Int. 2011;59:749-58
24. Prasad SS, Russell M, Nowakowska M. Neuroprotection induced in vitro by ischemic preconditioning and postconditioning: modulation of apoptosis and PI3K-Akt pathways. J Mol Neurosci. 2011;43:428-42
25. Zhang W, Miao Y, Zhou S, Jiang J, Luo Q, Qiu Y. Neuroprotective effects of ischemic postconditioning on global brain ischemia in rats through upregulation of hippocampal glutamine synthetase. J Clin Neurosci. 2011;18:685-689
26. Yuan Y, Guo Q, Ye Z, Pingping X, Wang N, Song Z. Ischemic postconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PI3K-Akt pathway. Brain Res. 2011;1367:85-93
27. Hyun DH, Lee M, Halliwell B, Jenner P. Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J Neurochem. 2003;86:363-73
28. Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678-99
29. Lehotský J, Murín R, Strapková A, Uríková A, Tatarková Z, Kaplán P. Time course of ischemia/reperfusion-induced oxidative modification of neural proteins in rat forebrain. Gen Physiol Biophys. 2004;23:401-15
30. Mathew SS, Bryant PW, Burch AD. Accumulation of oxidized proteins in Herpesvirus infected cells. Free Radic Biol Med. 2010;49:383-91
31. Norris EH, Giasson BI. Role of oxidative damage in protein aggregation associated with Parkinson's disease and related disorders. Antioxid Redox Signal. 2005;7:672-84
32. Li Z, Liu B, Yu J, Yang F, Luo Y, Ge P. Ischemic Postconditioning Rescues Brain Injury Caused by Focal Ischemia Reperfusion via Attenuating Proteins Oxidization. Journal of International Medical Research. 2012;40:954-66
33. Hochrainer K, Jackman K, Anrather J, Iadecola C. Reperfusion Rather than Ischemia Drives the Formation of Ubiquitin Aggregates After Middle Cerebral Artery Occlusion. Stroke. 2012 Jun 14
34. Ge P, Zhang F, Zhao J, Liu C, Sun L, Hu B. Protein degradation pathways after brain ischemia. Curr Drug Targets. 2012;13:159-65
35. Liu CL, Martone ME, Hu BR. Protein ubiquitination in postsynaptic densities after transient cerebral ischemia. J Cereb Blood Flow Metab. 2004;24:1219-25
36. Merker K, Sitte N, Grune T. Hydrogen peroxide-mediated protein oxidation in young and old human MRC-5 fibroblasts. Arch Biochem Biophys. 2000;375:50-4
37. McNaught KS, Björklund LM, Belizaire R, Isacson O, Jenner P, Olanow CW. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport. 2002;13:1437-41
38. Asai A, Tanahashi N, Qiu JH, Saito N, Chi S, Kawahara N, Tanaka K, Kirino T. Selective proteasomal dysfunction in the hippocampal CA1 region after transient forebrain ischemia. J Cereb Blood Flow Metab. 2002;22:705-10
39. Phillips JB, Williams AJ, Adams J, Elliott PJ, Tortella FC. Proteasome inhibitor PS519 reduces infarction and attenuates leukocyte infiltration in a rat model of focal cerebral ischaemia. Stroke. 2000;31:1686-93
40. Henninger N, Sicard KM, Bouley J, Fisher M, Stagliano NE. The proteasome inhibitor VELCADE reduces infarction in rat models of focal cerebral ischaemia. Neurosci Lett. 2006;398:300-5
41. Ge P, Zhang J, Wang X, Meng F, Li W, Luan Y, Ling F, Luo Y. Inhibition of autophagy induced by proteasome inhibitor increases cell death in human SHG-44 glioma cells. Acta Pharmacologica Sinica. 2009;30:1046-52
42. Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68-78
43. Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM Jr, Gottlieb RA. Autophagy Induced by Ischemic Preconditioning is Essential for Cardioprotection. J Cardiovasc Transl Res. 2010;3:365-373
44. Laskowska E, Matuszewska E, Kuczyńska-Wiśnik D. Small heat shock proteins and protein-misfolding diseases. Curr Pharm Biotechnol. 2010;11:146-57
Author contact
![]() Corresponding author: Pengfei Ge, Department of Neurosurgery, First Bethune Hospital of Jilin University, Changchun 130021, Jilin Province, P.R.China. E-mail: pengfeigecom, or gepfedu.cn.
Corresponding author: Pengfei Ge, Department of Neurosurgery, First Bethune Hospital of Jilin University, Changchun 130021, Jilin Province, P.R.China. E-mail: pengfeigecom, or gepfedu.cn.