Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Direct and indirect actions of...
Immune regulation of osteoclast...
Immune regulation of bone loss...
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2012; 9(9):825-832. doi:10.7150/ijms.5180 This issue Cite
Review
Immune Regulation of Osteoclast Function in Postmenopausal Osteoporosis: A Critical Interdisciplinary Perspective
Renqing Zhao ![]()
College of Physical Education and Health Sciences, Zhejiang Normal University, Zhejiang Jinhua, China.
Received 2012-9-7; Accepted 2012-10-15; Published 2012-10-27
Abstract
Extensive studies on cross talk between immune and skeletal systems in autoimmune diseases give rise to a new discipline of 'osteoimmunolgy', which explores the molecular regulation of osteoclasts by immune system. Postmenopausal osteoporosis is recognized as a cytokine driven disease, but the mechanism that how estrogen deficiency interplaying with cytokines to stimulate bone loss remains to be elucidated. Although the effect of individual cytokines on osteoclast formation is well characterized, the major challenge is to fit a multitude of redundant pathways and cytokines into a systemic model of postmenopausal osteoporosis. This review presents current findings and hypothesis to explain estrogen deficiency-stimulated bone loss in a critical interdisciplinary perspective. To better understand the interaction between osteoclasts and immune system in postmenopausal osteoporosis, many of the lessons have been explored in animal models.
Keywords: estrogen deficiency, osteoimmunology, cytokine, osteoclast.
Introduction
Women with natural menopause frequently suffer from osteoporosis, a systemic skeletal disorder characterized by reduced bone mineral density and increased fracture risk[1, 2]. This pathological process is heightened by rapid bone loss during the first five years of the onset of menopause.
Estrogen deficiency increases osteoclast formation by increasing haematopoietic progenitors and providing a larger recruited osteoclast progenitor pool[3-5]. The upregulated formation and activation of osteoclasts lead to cortical porosity and enlarged resorption areas in trabecular surfaces[1, 2]. In addition, estrogen depletion also increases the life span of osteoclasts and this event leads to prolonged bone loss, deeper resorption cavities and trabecular perforation increasing the fragility of bone[6]. This event contributes to a longer and slower period of bone wasting following acute phase of bone loss[2].
The bone loss is partly compensated by augmentation of bone formation due to the increased osteoblastogenesis. This event is fueled by increasing the number of mesenchymal progenitors capable of committing to the osteoblastic lineage and thus promotes proliferation of early osteoblast precursors[3, 7, 8]. The net increase of bone formation, however, is limited by increasing apoptosis of osteoblasts induced by estrogen deprivation[9, 10]. Therefore, the balance of bone remodeling tipped to the bone loss. However, the most elusive question is that the exact molecular mechanism of estrogen deficiency regulating bone absorption remains unclear.
Direct and indirect actions of estrogen deficiency on osteoclast formation
Since successful clone of estrogen receptors (ERs) ERα and ERβ, direct signaling pathways of estrogen regulating bone cells have extensively investigated. The classical nuclear transcription pathway begins with the binding estrogen with its receptors, and in turn the hormone-bound receptors dimerize and translocate into nucleus, in where this dimer binds to a specific DNA sequences called estrogen response elements (EREs) directly[11, 12]. This event results in increased or decreased mRNA levels associated with protein production[13, 14]. In the none-classical transcription pathway, the estrogen-ER dimer coactivate with other transcription factors (TFs) e.g. AP-1, NF-κB and SP-1, and binds non-ERE site to modulate gene expression by stimulating the general transcriptional machinery[15, 16]. Besides classical and non-classical nuclear pathways, an alternative nongenomic signaling is initiated by membrane bound receptors ERs and GPR30 acting as an acute response phase within seconds or minutes in bone cells[17, 18]. It is firmly established that estrogen receptors present in osteoblasts and osteoclasts can directly regulate their functions. Therefore, stimulating estrogen receptors can active osteoblast anabolic activities and depressed osteoclast catabolic activities.
However, a recent interesting study demonstrated that estrogen alone did not directly affect osteoclast precursors, but when osteoclast precursors were cultured with osteoblastic cells estrogen had the capacity to inhibit osteoclast formation[19]. This indicated that indirect pathway may play a more important role in regulating the effect of estrogen on osteoclast development. Indeed, numerous evidences have demonstrated that osteoblast together with other bone marrow stromal cells secret essential cytokines and growth factors to drive the commitment of haematopoietic progenitors to osteoclastic lineage and thus the increment of osteoclast formation[20, 21], and estrogen deficiency induced bone loss by up regulating cytokine production in immune cells[22]. Therefore, the mechanism that estrogen deficiency induced bone loss seems more complicated and interplay between estrogen deficiency and immune cells may play a pivotal role in regulating bone absorption in postmenopausal osteoporosis.
The close relationship between immune and skeletal systems should not be surprising, because there exist in many similar receptors, transcription factors, and coactivators between the two systems. After the discovery of RANKL/RANK signaling pathway in bone cells, the connection between immune system and bone firmly acquires the molecular supports.
Immune regulation of osteoclast formation
Both immune cells of peripheral blood and osteoclasts of bone arise from pluripotent hematopoietic stem cells (HSCs) that reside in bone marrow after birth. HSCs have the capacity for self-renewal and produce daughter cells which restrictively develop into respective cell lineage. HSCs give rise to the multipotent progenitors (MPPs) which lacking the capacity of self-renewal (Figure 1). MPPs further differentiate into two distinct lineage populations, termed common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs) [23-25]. CMPs subsequently differentiate into granulocyte-macrophage progenitors (GMPs) which give rise to osteoclasts, macrophages, granulocytes and dendritic cells[26, 27]. CLPs develop into T, B and natural killer cells[26, 27]. Therefore, it is at MPP stage that cells regulating adaptive immunity and osteoclasts share a common progenitor, and essential commitments are made that decide whether immune cells or osteoclastic cells will be produced.
Hematopoietic stem cell differentiation. All hematopoietic cells circulating in peripheral blood arise from pluripotent hematopoietic stem cells which differentiate into multipotent progenitors (MPPs). At the stage of multipotent progenitors (MPPs), important differentiation occurs. MPPs differentiate into the common myeloid progenitors (CMPs) and further granulocyte-macrophage progenitors (GMPs) which give rise to osteoclasts, macrophages, granulocytes and dendritic cells. MPPs also differentiate into common lymphoid progenitors (CLP) which develop into T, B and natural killer cells.
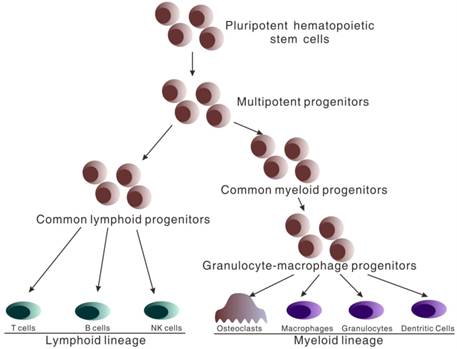
It is reasonable to assume that sharing the same progenitors and similar development environment, immune cells and osteoclasts may also share some similar physiological function. This presumption is supported by recent studies which suggest that immune cells and osteoclasts share a number of regulatory molecules, including cytokines, receptors, signaling pathway, growth factors, and transcription factors[20, 28-30]. Therefore, cytokines that produced by immune cells and regulate adaptive response may also affect osteoclast function through the common receptors, signaling pathway, and transcription factors. This hypothesis is supported by the evidences learned from experimental animal models and human observations with autoimmune diseases[31-37].
Giving the facts that bone marrow stromal cells secret or express cytokines and growth factors supporting the development of osteoclast and immune cells, it indicates that there exists in cross talk between immune and skeletal systems. The discovery that RANKL/RANK signaling pathway plays a pivotal role both in adaptive immunity and osteoclastogenesis provides firm molecular evidence to interconnect immune and skeletal systems.
Receptor activator of nuclear factor κ-B ligand (RANKL), known as tumor necrosis factor ligand superfamily member 11 (TNFSF11), originally was found as a functional molecule involved in the regulation of T cell-dependent immune response. Recently, it is found that RANKL also plays an important role in regulating osteoclast development[29, 30]. Osteoblasts and their precursors express RANKL both in membrane-bound and soluble forms. RANKL binds to the tansmembrane receptor RANK (receptor activator of nuclear factor κ-B) expressed on the surface of osteoclasts and their precursors. It promotes proliferation and differentiation of osteoclast precursors, and the maturation and activity of osteoclasts. The osteoclastogenic activity induced by RANKL-RANKL binding is inhibited by another member of the TNF receptor superfamily 'osteoprotegerin' (OPG) produced by osteoblasts. Therefore, OPG, by preventing RANKL-RANK binding, functions as a potent anti-osteoclastogenic cytokine.
Osteoclast formation needs the presence of another essential cytokine, macrophage colony stimulating factor (M-CSF) produced by several cell types including osteoblasts, stromal cells. M-CSF activates an intracellular cascade upon binding its receptor, the c-fms expressed at the surface of osteoclastic cells, and leads to proliferation and differentiation of osteoclast precursors and survival of mature osteoclasts[30].
Although RANKL and M-CSF are essential for osteoclastogenesis, additional cytokines contribute to the complex regulation of osteoclast formation both in physiological and pathological condition such as estrogen deficiency and inflammation. Attesting to the key roles of TNF, IL-1, IL-6 and IL-7 in the estrogen deficiency-induced bone loss[38-42], it indicates that bone remodeling is regulated by a large number of cytokines which cooperate and overlap effects on osteoclastogenesis. Although the exact mechanisms of cytokine driven bone loss in postmenopausal osteoporosis are not well defined, recent developments learned from both animal and human studies help us get a deeper understanding.
Immune regulation of bone loss induced by estrogen deficiency
T cells and estrogen deficiency-induced bone loss
Despite of several inverse reports[43, 44], the main body of literatures firmly supports the essential role of activated T cells in regulating bone loss induced by estrogen deficiency[38, 45-50]. The core evidences have been learned both from experimental animal studies and human observations. First, T cell-deficient nude mice and T cell-deleted wild mice failed to response to ovariectomy contrasted to their control littermates that experienced deleterious bone loss and microarchitecture damage[46-50]. Second, the capacity of ovariectomy to Induce bone absorption was restored when reconstitution of nude mice with T cells from wild type mice[45]. In addition, while menopause up regulated T cell activity and increased T cell production of cytokines[37], hormone therapy decreased osteoclastogenic cytokine production in postmenopausal women[51].
Although up regulated T cell activity and cytokine production play a pivotal role in estrogen deficiency-induced bone loss, the source and characteristics of T cells remains to be defined. Several papers reported that the expansion of bone marrow T cell populations and increment of cytokine productions, especially TNF, increased osteoclastogenesis in ovariectomy mice[38, 39]. The dominant output of this cytokine is a net crease in RANKL production. Increment of TNF act synergistically with RANKL and thus increase commitment of hematopoietic pluripotent progenitors to osteoclastic lineage and thus expand the osteoclast precursor pool and ultimately the osteoclast formation.
Thymus not only contributes to the output of naïve T cells into peripheral blood but regulates the production, mobilization and recruitment of hematogenous thymocyte progenitors in bone marrow[52]. Therefore, increased thymic output induced by estrogen deficiency both affects peripheral blood T cell populations and the expansion of bone marrow T cell pool. Cenci et al [38] demonstrated that while ovariectomy decreases the BMD by 30% in euthymic mice, nude mice completely protected against ovariectomy induced bone loss. It indicates that the enhanced thymic output of T cells plays a previously unrecognized role in the estrogen deficiency-induced bone loss. Ryan et al [53] reported that ovariectomy increased thymic T cell output and the expansion of bone marrow T cell populations. In fact, recent studies indicated that thymus regulates the formation and release of hematogenous thymocyte progenitors in bone marrow through thymus-bone marrow feedback loop.[52].Therefore, estrogen deficiency-increased thymic output both affects systemic bone remodeling and osteoclastogenesis in bone marrow.
TNF and Estrogen deficiency-stimulated bone loss
Activated T cells play a pivotal role in regulating estrogen deficiency-driven osteoclast formation, because T cells have the capacity to secrete a wide repertoire of osteoclastogenic cytokines. One of such responsible cytokines is TNF, a cytokine that increases osteoclast formation by stimulating bone marrow stromal cell production of RANKL and M-CSF, and by promoting the responsiveness of osteoclast precursors to RANKL[35]. The relevance of TNF in up-regulating osteoclast formation during estrogen deficiency has been demonstrated by multiple animal models. Mice with TNF-/- or absence of the p55 TNF receptor were unable to induce bone loss after ovary surgically removed[45]. Furthermore, either insensitive to TNF due to overexpression of a soluble TNF receptor or treatment with TNF inhibitor protects from ovariectomy-stimulated bone loss[54, 55]. Elevated level of TNF was found in the bone marrow of ovariectomy mice[38] and in the conditioned media of peripheral blood cells of postmenopausal women[37]. It appears that upregulated production of TNF by activated T cells is essential for estrogen deficiency-induced bone loss.
The major focus is to understand how estrogen deficiency up-regulates T cell production of TNF. In a subsequent paper, Pacifici group showed that it was the result of a complex pathway involved in the bone marrow and thymus. Estrogen deficiency stimulates the expression of MHCII in macrophages and dendritic cells, and subsequently expands the proliferation and lifespan of bone marrow T cells[47, 56]. This process is regulated by a mechanism that estrogen deficiency stimulates the bone marrow macrophage expression of the gene encoding Class II Transactivator (CIITA), a transcriptional coactivator at the MHC II promoter[56]. One of the cytokines responsible for un-regulating CIITA expression in macrophages is INF. The ability of INF activating antigen presenting cells was demonstrated by the facts that ovariectomy fails to induce bone loss in mice with INF receptor knockout[56]. Estrogen deficiency down regulating TGFβ production partly contributes to increased INF production[46] and up regulating production of IL-7[39, 53] in the condition of ovariectomy bears another mechanism of enhanced expression of TNF and INF.
IL-6 and estrogen deficiency-induced bone loss
Another cytokine responsible for estrogen deficiency-induced bone loss is IL-6, a member of the gp130 cytokine family. IL-6 is widely recognized as a potent stimulator of osteoclast-driven bone absorption in the context of chronic inflammation and estrogen deficiency[57]. Experimental animal studies suggests that transgenic mice, overexpressing IL-6, exhibit severe impairment of cortical and trabecular bone microarchitecture, together with increased osteoclastogenesis and decreased osteoblastogenesis[58]. Estrogen has the capacity of inhibiting the production of IL-6 in osteoblastic linage cells[59, 60] and neutralizing IL-6 function by antibody prevents mice from ovariectomy-induced bone loss[5]. Estrogen depletion causes in wild type animals significant bone loss together with increased bone turnover rates, but ovariectomy failed to induce any changes in both bone mass and bone remodeling rates in the IL-6 deficient mice[42]. Molecular studies indicate that bone samples from postmenopausal women with osteoporosis expressed more IL-6 mRNA than postmenopausal women with normal BMD[61]. Moreover, IL-6 production by stimulated peripheral blood cells was significantly increased in postmenopausal osteoporosis and level of IL-6 negatively related to lumber BMD[41].
Although the main sources of IL-6 in skeletal system are osteoblastic cells and stromal cells, the dominant effect of IL-6 on bone is on osteoclastic cells and bone resorption[62]. Attesting to the indirect mechanism of IL-6 stimulating osteoclast formation is reports by Hattersley that IL-6 stimulated osteoclastogenesis when co-cultured with osteoblasts but had not effects in a highly purified osteoclasts culture[63]. Further function studies indicated that receptors expressed on osteoblasts but not on osteoclasts were required for IL-6 to regulate osteoclastogenesis, though higher level of IL-6 receptors are found on osteoclastic cells[64]. IL-6 firstly binds to IL-6R and forms complex with gp130 to stimulate intracellular signaling machinery. The main effect of IL-6 is to stimulate osteoblastic downstream production of signaling molecules especially RANKL, which subsequently enhances osteoclast formation and activity[65]. Furthermore, the activity of IL-6 on osteoclasts frequently interplays with IL-1 and TNF, and IL-6 increases the stimulatory effect of IL-1 and TNF on bone resorption by increasing osteoclastic progenitor pool[66, 67]. As IL-6 is indispensable for the differentiation from naïve CD4+ T cells to Th17 cells, a potent osteoclastogenic inducer in the condition of chronic inflammation and autoimmune diseases, therefore new mechanism of IL-6 regulating osteoclastogenesis need to be defined.
IL-7 and estrogen deficiency-induced bone loss
IL-7 is a potent osteoclastogenic cytokine initially recognized as a growth and homeostasis factor for T and B cells. Recent evidence demonstrates that IL-7 is one of the indispensable cytokines that mediating estrogen deficiency stimulated bone loss[68, 69]. IL-7 production is up regulated by estrogen deficiency in bone marrow and increased IL-7 levels contribute to bone absorption[39, 53]. Weitzmann and colleagues[39] reports that ovariectomy increases IL-7 mRNA levels by threefold and IL-7 protein concentrations by 35% in bone marrow compared with sham-operated and estrogen-replete controls. Furthermore, neutralization of IL-7 with specific antibody (M25) completely protects from bone loss by downregulating bone resorption and stimulating bone formation. Ryan et al [53]further demonstrates that the thymus, spleen and bone marrow are the all lymphoid source of IL-7 in response to estrogen deficiency.
IL-7 increases bone loss mainly by increasing RANKL and TNF producing T cells. Recently, it is showed that systemic administration of IL-7 significantly enhanced osteoclast formation in human peripheral blood cells by increasing osteoclastogenic cytokine production in T cells,[70] and IL-7 failed to stimulate bone loss in T cell deficient nude mice[71]. When reconstitution of nude mice with T cells, IL-7 administration restored the capacity of ovariectomy to induce osteoclastic bone absorption, concurrent with increased production of RANKL and TNF in splenic T cells[71]. Ovariectomy significantly promotes early stages of T cell development, and this effect is inhibited in part by anti-IL-7 antibody[53]. Those data suggested that estrogen deficiency induces bone loss through an IL-7 dependent mechanism that contributes to the expansion of T cell populations.
Despite great progress, the exact mechanism, by which IL-7 regulates estrogen deficiency caused expansion of T cell population, is far from defined. Several explanations help us better understand the mechanism. First, as T cells express IL-7R, estrogen deficiency stimulated IL-7 expression directly enhances T cell proliferation and activity by lowering tolerance to weak self antigens[21]. Second, attesting to evidence that IL-7 has the capacity to promote antigen presentation and thus T cell activation through stimulating INF production,[72] it assumes that there exists in indirect action. Recently, Ryan et al[53] reported that ovariectomy causes significant increase in number of INF producing T cells and increment of macrophage CIITA expression and macrophage antigen presentation, which phenomena were completely prevented by administration of IL-7 antibody. Finally, attesting to the findings that ovariectomy induced osteoclastogenesis in a population of B220+ cells and IL-7 is best known to promote differentiation and survival of B220+ cells[73, 74], it is reasonable to assume that IL-7 mediates the estrogen deficiency stimulated bone loss by a mechanism related to B220+ cells. Evidence supporting this idea showed that IL-7 induced expansion of B220+ cells is sufficient to induce bone loss[71].
Interaction of osteoclastogenic cytokines in estrogen deficiency-induced bone loss
Although individual cytokine playing a key role in regulating estrogen deficiency-induced bone loss is well investigated, these proinflammatory cytokines working in a synergic fashion remain unclear. TNF induces osteoclastic bone absorption only in the presence of IL-1, and TNF together with IL-1 may play an important role in bone resorption[75]. IL-1 is a pleiotropic cytokine and induces the expression of a large variety of proinflammatory cytokines including IL-6[76]. Indeed, effect of IL-6 on osteoclastogenesis is interconnected with IL-1 and TNF. On the one hand, IL-6 stimulates osteoclast formation by inducing IL-1 release and administration of anti-IL-1 inhibits IL-6 stimulated osteoclastogenesis[66]. On the other hand, IL-6 mediates the effects of TNF on osteoclastic cell formation[67]. IL-7 is best recognized for stimulating production of TNF in T cells. However, both IL-7 and IL-7Rα expressions are up regulated by TNF[77, 78]. Therefore, an interplay mechanism may exist in between TNF and IL-7.
Conclusions
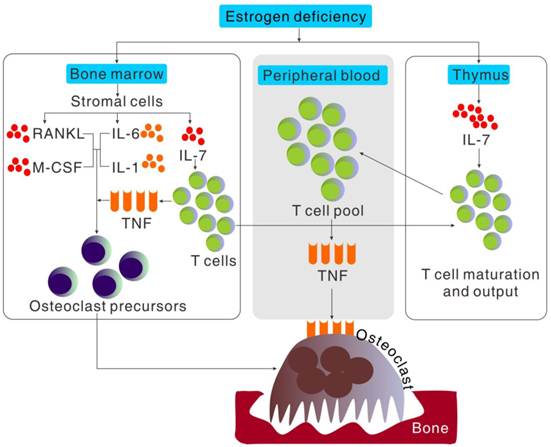
Accumulating evidences both from ovariectomized animal studies and postmenopausal women observation strongly suggested that estrogen deficiency-induced bone loss is a complex interplay of estrogen, the immune system and bone rather than an action of estrogen directly on bone cells (Figure 2). This greatly helps us better understand postmenopausal osteoporosis and may provide new therapy for this disease. In the past two decades, the effect of single cytokine produced by activated T cells or bone marrow stromal cells on osteoclast formation has been extensively investigated, and new anti-cytokine therapy for treating postmenopausal osteoporosis demonstrate successful in alleviating osteoporosis syndrome and lowering fracture accidents. However, increasing evidences suggested that the postmenopausal osteoporosis is a systemic model of integrated pathway and cytokines working in a cooperative fashion. Especially with the discovery of a new CD4+ T cell subset and its signature cytokine, named Th17 cells and IL-17, not only the Th1/Th2 paradigm but the nature and source of those pro-inflammatory cytokines need to be redefined.
Cytokine regulation of osteoclast formation induced by estrogen deficiency. Bone loss induced by estrogen deficiency is a complex effect of a multitude of redundant pathways and cytokines working in a cooperative fashion to regulate osteoclastogenesis. Estrogen deficiency causes in a global increment in IL-7 production, especially in thymus, bone marrow and spleen. The increased production of IL-7 in bone marrow increases T cell populations and activity which increases TNF production and importation of hematogenous thymocyte progenitors into thymus. The increment of IL-7 in thymus increases the output of naïve CD4+T cell output into peripheral blood and in turn the expansion of T cell pool which increased the production of TNF. Estrogen deficiency also increases the stromal cell production of cytokines such as IL-6, IL-1, RANKL and M-CSF, that promote proliferation and differentiation of osteoclast precursors independently or cooperatively in bone marrow.
Acknowledgements
This study was based upon work funded by Zhejiang Provincial Natural Science Foundation of China under Grant (No. Y2110954).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mcnamara L M. Perspective on post-menopausal osteoporosis: establishing an interdisciplinary understanding of the sequence of events from the molecular level to whole bone fractures. J R Soc Interface. 2010;7(44):353-72
2. Lerner U H. Bone remodeling in post-menopausal osteoporosis. J Dent Res. 2006;85(7):584-95
3. Rosen C J. Pathogenesis of osteoporosis. Baillieres Best Pract Res Clin Endocrinol Metab. 2000;14(2):181-93
4. Jilka R L, Passeri G, Girasole G. et al. Estrogen loss upregulates hematopoiesis in the mouse: a mediating role of IL-6. Exp Hematol. 1995;23(6):500-6
5. Jilka R L, Hangoc G, Girasole G. et al. Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science. 1992;257(5066):88-91
6. Bell K, Loveridge N, Lunt M. et al. Oestrogen suppression increases Haversian resorption depth as well as remodelling activity in women with endometriosis. Bone. 1996;19:131S
7. Di Gregorio G B, Yamamoto M, Ali a A. et al. Attenuation of the self-renewal of transit-amplifying osteoblast progenitors in the murine bone marrow by 17 beta-estradiol. J Clin Invest. 2001;107(7):803-12
8. Jilka R L, Takahashi K, Munshi M. et al. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow. Evidence for autonomy from factors released during bone resorption. J Clin Invest. 1998;101(9):1942-50
9. Kousteni S, Han L, Chen J R. et al. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. 2003;111(11):1651-64
10. Kousteni S, Bellido T, Plotkin L I. et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104(5):719-30
11. Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor alpha and beta: impact on human health. Mol Aspects Med. 2006;27(4):299-402
12. Nilsson S, Makela S, Treuter E. et al. Mechanisms of estrogen action. Physiol Rev. 2001;81(4):1535-65
13. Smith C L, O'malley B W. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25(1):45-71
14. Tsai M J, O'malley B W. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451-86
15. Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett. 2006;238(1):1-14
16. Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001;62:231-52
17. Levin E R. Cellular functions of plasma membrane estrogen receptors. Steroids. 2002;67(6):471-5
18. Kelly M J, Levin E R. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab. 2001;12(4):152-6
19. Michael H, Harkonen P L, Vaananen H K. et al. Estrogen and testosterone use different cellular pathways to inhibit osteoclastogenesis and bone resorption. J Bone Miner Res. 2005;20(12):2224-32
20. Nakashima T, Takayanagi H. Osteoimmunology: crosstalk between the immune and bone systems. J Clin Immunol. 2009;29(5):555-67
21. Pacifici R. The immune system and bone. Arch Biochem Biophys. 2010;503(1):41-53
22. Breuil V, Ticchioni M, Testa J. et al. Immune changes in post-menopausal osteoporosis: the Immunos study. Osteoporos Int. 2010;21(5):805-14
23. Adolfsson J, Borge O J, Bryder D. et al. Upregulation of Flt3 expression within the bone marrow Lin(-)Sca1(+)c-kit(+) stem cell compartment is accompanied by loss of self-renewal capacity. Immunity. 2001;15(4):659-69
24. Akashi K, Traver D, Miyamoto T. et al. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404(6774):193-7
25. Kondo M, Weissman I L, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91(5):661-72
26. Mandel E M, Grosschedl R. Transcription control of early B cell differentiation. Curr Opin Immunol. 2010;22(2):161-7
27. Kondo M, Wagers a J, Manz M G. et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21:759-806
28. Nakashima T, Takayanagi H. Osteoclasts and the immune system. J Bone Miner Metab. 2009;27(5):519-29
29. Caetano-Lopes J, Canhao H, Fonseca J E. Osteoimmunology--the hidden immune regulation of bone. Autoimmun Rev. 2009;8(3):250-5
30. Quinn J M, Saleh H. Modulation of osteoclast function in bone by the immune system. Mol Cell Endocrinol. 2009;310(1-2):40-51
31. Okamoto K, Takayanagi H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther. 2011;13(3):219
32. Islander U, Jochems C, Lagerquist M K. et al. Estrogens in rheumatoid arthritis; the immune system and bone. Mol Cell Endocrinol. 2011;335(1):14-29
33. Maruotti N, Grano M, Colucci S. et al. Osteoclastogenesis and arthritis. Clin Exp Med. 2011;11(3):137-45
34. Karmakar S, Kay J, Gravallese E M. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum Dis Clin North Am. 2010;36(2):385-404
35. Pacifici R. T cells: critical bone regulators in health and disease. Bone. 2010;47(3):461-71
36. Kosa J P, Balla B, Kiss J. et al. Postmenopausal expression changes of immune system-related genes in human bone tissue. J Clin Immunol. 2009;29(6):761-8
37. D'amelio P, Grimaldi A, Di Bella S. et al. Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: a key mechanism in osteoporosis. Bone. 2008;43(1):92-100
38. Cenci S, Weitzmann M N, Roggia C. et al. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. J Clin Invest. 2000;106(10):1229-37
39. Weitzmann M N, Roggia C, Toraldo G. et al. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110(11):1643-50
40. Wei S, Kitaura H, Zhou P. et al. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005;115(2):282-90
41. Zheng S X, Vrindts Y, Lopez M. et al. Increase in cytokine production (IL-1 beta, IL-6, TNF-alpha but not IFN-gamma, GM-CSF or LIF) by stimulated whole blood cells in postmenopausal osteoporosis. Maturitas. 1997;26(1):63-71
42. Poli V, Balena R, Fattori E. et al. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. Embo J. 1994;13(5):1189-96
43. Lee S K, Kadono Y, Okada F. et al. T lymphocyte-deficient mice lose trabecular bone mass with ovariectomy. J Bone Miner Res. 2006;21(11):1704-12
44. Anginot A, Dacquin R, Mazzorana M. et al. Lymphocytes and the Dap12 adaptor are key regulators of osteoclast activation associated with gonadal failure. PLoS One. 2007;2(7):e585
45. Roggia C, Gao Y, Cenci S. et al. Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci U S A. 2001;98(24):13960-5
46. Gao Y, Qian W P, Dark K. et al. Estrogen prevents bone loss through transforming growth factor beta signaling in T cells. Proc Natl Acad Sci U S A. 2004;101(47):16618-23
47. Gao Y, Grassi F, Ryan M R. et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117(1):122-32
48. Grassi F, Tell G, Robbie-Ryan M. et al. Oxidative stress causes bone loss in estrogen-deficient mice through enhanced bone marrow dendritic cell activation. Proc Natl Acad Sci U S A. 2007;104(38):15087-92
49. Li J Y, Tawfeek H, Bedi B. et al. Ovariectomy disregulates osteoblast and osteoclast formation through the T-cell receptor CD40 ligand. Proc Natl Acad Sci U S A. 2011;108(2):768-73
50. Robbie-Ryan M, Pacifici R, Weitzmann M N. IL-7 drives T cell-mediated bone loss following ovariectomy. Ann N Y Acad Sci. 2006;1068:348-51
51. Rogers A, Eastell R. The effect of 17beta-estradiol on production of cytokines in cultures of peripheral blood. Bone. 2001;29(1):30-4
52. Goldschneider I. Cyclical mobilization and gated importation of thymocyte progenitors in the adult mouse: evidence for a thymus-bone marrow feedback loop. Immunol Rev. 2006;209:58-75
53. Ryan M R, Shepherd R, Leavey J K. et al. An IL-7-dependent rebound in thymic T cell output contributes to the bone loss induced by estrogen deficiency. Proc Natl Acad Sci U S A. 2005;102(46):16735-40
54. Kimble R B, Bain S, Pacifici R. The functional block of TNF but not of IL-6 prevents bone loss in ovariectomized mice. J Bone Miner Res. 1997;12(6):935-41
55. Ammann P, Rizzoli R, Bonjour J P. et al. Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiency. J Clin Invest. 1997;99(7):1699-703
56. Cenci S, Toraldo G, Weitzmann M N. et al. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc Natl Acad Sci U S A. 2003;100(18):10405-10
57. Edwards C J, Williams E. The role of interleukin-6 in rheumatoid arthritis-associated osteoporosis. Osteoporos Int. 2010;21(8):1287-93
58. De Benedetti F, Rucci N, Del Fattore A. et al. Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006;54(11):3551-63
59. Girasole G, Jilka R L, Passeri G. et al. 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J Clin Invest. 1992;89(3):883-91
60. Stein B, Yang M X. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Mol Cell Biol. 1995;15(9):4971-9
61. Ralston S H. Analysis of gene expression in human bone biopsies by polymerase chain reaction: evidence for enhanced cytokine expression in postmenopausal osteoporosis. J Bone Miner Res. 1994;9(6):883-90
62. Holt I, Davie M W, Marshall M J. Osteoclasts are not the major source of interleukin-6 in mouse parietal bones. Bone. 1996;18(3):221-6
63. Hattersley G, Dorey E, Horton M A. et al. Human macrophage colony-stimulating factor inhibits bone resorption by osteoclasts disaggregated from rat bone. J Cell Physiol. 1988;137(1):199-203
64. Udagawa N, Takahashi N, Katagiri T. et al. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J Exp Med. 1995;182(5):1461-8
65. Bellido T, Stahl N, Farruggella T J. et al. Detection of receptors for interleukin-6, interleukin-11, leukemia inhibitory factor, oncostatin M, and ciliary neurotrophic factor in bone marrow stromal/osteoblastic cells. J Clin Invest. 1996;97(2):431-7
66. Kurihara N, Bertolini D, Suda T. et al. IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J Immunol. 1990;144(11):4226-30
67. Devlin R D, Reddy S V, Savino R. et al. IL-6 mediates the effects of IL-1 or TNF, but not PTHrP or 1,25(OH)2D3, on osteoclast-like cell formation in normal human bone marrow cultures. J Bone Miner Res. 1998;13(3):393-9
68. Ross F P. Interleukin 7 and estrogen-induced bone loss. Trends Endocrinol Metab. 2003;14(4):147-9
69. Lee S K, Surh C D. Role of interleukin-7 in bone and T-cell homeostasis. Immunol Rev. 2005;208:169-80
70. Weitzmann M N, Cenci S, Rifas L. et al. Interleukin-7 stimulates osteoclast formation by up-regulating the T-cell production of soluble osteoclastogenic cytokines. Blood. 2000;96(5):1873-8
71. Toraldo G, Roggia C, Qian W P. et al. IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor kappa B ligand and tumor necrosis factor alpha from T cells. Proc Natl Acad Sci U S A. 2003;100(1):125-30
72. Borger P, Kauffman H F, Postma D S. et al. IL-7 differentially modulates the expression of IFN-gamma and IL-4 in activated human T lymphocytes by transcriptional and post-transcriptional mechanisms. J Immunol. 1996;156(4):1333-8
73. Katavic V, Grcevic D, Lee S K. et al. The surface antigen CD45R identifies a population of estrogen-regulated murine marrow cells that contain osteoclast precursors. Bone. 2003;32(6):581-90
74. Sato T, Shibata T, Ikeda K. et al. Generation of bone-resorbing osteoclasts from B220+ cells: its role in accelerated osteoclastogenesis due to estrogen deficiency. J Bone Miner Res. 2001;16(12):2215-21
75. Kobayashi K, Takahashi N, Jimi E. et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191(2):275-86
76. Dinarello C A. The interleukin-1 family: 10 years of discovery. Faseb J. 1994;8(15):1314-25
77. Harada S, Yamamura M, Okamoto H. et al. Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. 1999;42(7):1508-16
78. Tian B, Nowak D E, Jamaluddin M. et al. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem. 2005;280(17):17435-48
Author contact
![]() Corresponding author: Renqing Zhao, College of Physical Education and Health Sciences, Zhejiang Normal University, 688 Yingbin Road, Jinhua, Zhejiang Province, 321004 China. Tel: +86(579) 8229-8809 Fax: +86(579)8229-7990 Email: renzhaocn.
Corresponding author: Renqing Zhao, College of Physical Education and Health Sciences, Zhejiang Normal University, 688 Yingbin Road, Jinhua, Zhejiang Province, 321004 China. Tel: +86(579) 8229-8809 Fax: +86(579)8229-7990 Email: renzhaocn.