International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2012; 9(2):157-162. doi:10.7150/ijms.3880 This issue Cite
Research Paper
Inhaled Nitric Oxide Therapy Fails to Improve Outcome in Experimental Severe Influenza
Ilyse Darwish1,2, Chris Miller3, Kevin C. Kain1,2,4, W. Conrad Liles1,2,4 ![]()
1. Institute of Medical Science, University of Toronto, Toronto, Canada;
2. Sandra A. Rotman Laboratories, McLaughlin-Rotman Centre for Global Health, University Health Network-Toronto General Hospital, University of Toronto, Toronto;
3. Division of Respiratory Medicine, Department of Medicine, University of British Columbia, Vancouver, Canada;
4. Division of Infectious Diseases, Department of Medicine, University of Toronto, Toronto, Canada.
Received 2011-11-29; Accepted 2012-1-6; Published 2012-1-13
Citation:
Darwish I, Miller C, Kain KC, Liles WC. Inhaled Nitric Oxide Therapy Fails to Improve Outcome in Experimental Severe Influenza. Int J Med Sci 2012; 9(2):157-162. doi:10.7150/ijms.3880. https://www.medsci.org/v09p0157.htm
Other stylesAbstract
In vitro, nitric oxide (NO) has been shown to have antimicrobial activity against a wide range of viruses, including influenza A virus. Therefore, we hypothesized that inhaled nitric oxide (iNO) would increase survival in vivo by reducing the viral load in C57Bl/6 mice infected with a lethal dose of influenza A/WSN/33 (H1N1; WSN/33) virus. NO was delivered to influenza-infected mice either continuously or intermittently at 80 or 160 ppm, respectively, using both prophylactic and post-infection treatment strategies. Murine survival and weight loss were assessed, and lung viral load was quantified via plaque assay. Here, we report that iNO administered prophylactically or post-influenza infection failed to improve survival of infected mice. No difference in lung viral load was observed between experimental groups. Although NO has antiviral activity against influenza A virus in vitro, iNO therapy provided no apparent benefit when used for treatment of influenza A virus infection in vivo.
Keywords: nitric oxide, severe influenza, influenza A/WSN/33
INTRODUCTION
Influenza A viruses infect approximately 5-15% of the population, resulting in 250-500 thousands deaths each year (1). The most widely used class of drugs for treatment of clinical influenza is the neuraminidase inhibitors, including oseltamivir and zanamivir. The clinical impact of these drugs is limited by the development of antiviral drug resistance. Specifically, decreased efficacy of neuraminidase inhibitors has been reported against seasonal H1N1 influenza and 2009 novel swine-origin H1N1 influenza, as well as avian influenza H5N1 virus (2-9). In addition, initiation of antiviral therapy in influenza A virus-infected individuals beyond the first 48-72 hours after the onset of influenza symptoms is associated with greater mortality and decreased antiviral efficacy compared with treatment initiated within 48-72 hours of symptom onset (10-15). These caveats underscore the need to develop novel and effective influenza therapeutic strategies. Further investigation of other intervention strategies which have shown promising results against influenza A viruses in vitro but have not been investigated in vivo are warranted.
Nitric oxide (NO) is an important cellular signalling molecule synthesized from L-arginine by NO synthase (NOS). There are three types of NOS: constituent and calcium-dependant isoforms that are principally present in endothelial and neuronal cells (eNOS and nNOS, respectively), and the inducible or calcium-independent isoform, iNOS (16). In the airways, NOS is present in a variety of cells, including macrophages, vascular endothelial cells, airway epithelial cells and neurons where NOS activity is known to mediate neurotransmission, smooth muscle contraction and mucin secretions. NO is also a well known biological mediator in the host response to infection (16, 17). Various inflammatory stimuli such as LPS and cytokines including IFNg and TNF can cause high and sustained NO production by iNOS; depending on the species, strain, infection dose and pathogen entry route, iNOS activity can result in pro- or anti-inflammatory responses, cytotoxicity, or cytoprotection [reviewed in (16)].
In vitro, NO antimicrobial activity has been demonstrated against a variety of viruses including ectromilia virus, vaccinia virus, herpes simplex type 1 viruses, coronavirus, and influenza A and B viruses (18-22). In these studies, administration of the NO donor S-nitroso-N-acetylpenicillamine (SNAP) to virus-infected cells significantly reduced viral burden. A human trial for treatment of severe acute respiratory syndrome (SARS) found inhaled NO (iNO), at 30 ppm or less, decreased the spread and intensity of lung infiltrates and improved arterial oxygen saturation (23).
Severe cases of influenza infection are often associated with multisystem organ failure and hypoxemic respiratory failure, including acute lung injury/acute respiratory distress syndrome (ALI/ARDS) requiring advanced mechanical ventilatory support (24, 25). Affected individuals may receive 'rescue' therapies, including iNO, in an attempt to improve outcome (25). However, iNO administration for ARDS secondary to viral pneumonia has not been specifically reported to improve clinical outcome (24, 25).
The objective of this study was to determine whether iNO administration could reduce viral load and improve survival in a murine model of severe influenza. Inhaled NO delivery would provide a safer and easier delivery method rather than administration of NO donors, as iNO is approved for treating term and near-term neonates with hypoxemic respiratory failure up to a dose of 80 parts per million (ppm) (26, 27). It has been reported that exogenous gaseous NO (gNO) at a high dose of no less than 160 ppm and with five hours of continuous exposure, can elicit a non-specific antimicrobial response against a broad range of microorganisms in vitro (28). [15] B. McMullin, D. Chittock, D. Roscoe, H. Garcha, L. Wang and C. Miller, The antimicrobial effect of nitric oxide on the bacteria that cause nosocomial pneumonia in mechanically ventilated patients in the icu. Resp. Care, 50 11 (2005), pp. 1451-1456. | View Record in Scopus |. In vivo, 160 ppm iNO treatment would result in NO binding to hemoglobin to form methemoglobin, resulting in reduced oxygen transport and hypoxemia, as well as the potential for elevated levels of the harmful NO metabolite NO2. However, Miller et al. (29) has shown that gNO in an intermittent delivery regimen of 160 ppm for 30 min every 3.5 hours can prevent methemoglobinemia and reduce the potential of host cell toxicity in vitro and in vivo (Miller C, personal communication), while retaining antimicrobial properties in vitro.
RESULTS
Continuous iNO at 80 ppm decreased survival and intermittent high dose iNO at 160 ppm did not increase survival of influenza A virus-infected mice
We evaluated the ability of iNO to improve survival of influenza A/WSN/33 (mouse-adapted H1N1 strain; WSN/33) infected mice. Experimental C57Bl/6 mice were inoculated intranasally with an 80-100% lethal dose of WSN/33 (1000 PFU). At 5 days post-infection, the majority of mice in all experimental groups experienced weight loss (Fig. 1a and 2a). At 7 days post-infection, mice began to reach euthanasia criteria (≤80% of day 0 weight), and by day 10 post-infection, most mice were euthanized (Fig. 1b and 2b). If 20% weight loss was not met by day 10 post-infection, the infection typically resolved, and surviving mice gained weight.
Weight loss over the course of infection was accelerated in mice administered continuous iNO at 80 ppm starting 1 hour prior to inoculation compared to infected control mice receiving compressed room air (P < 0.001) (Fig. 1a). Continuous iNO administration at 80 ppm starting 1 hour prior to inoculation significantly decreased survival of WSN/33-infected mice compared to infected control mice administered compressed room air (P < 0.01). During the course of infection, 100% of continuous iNO treated mice were euthanized compared to 80% of infected control mice (Fig. 1b). Intermittent iNO administration at 160 ppm for 30 min intervals every 3.5 hours starting either 1 hour prior to or 4 hours post-infection resulted in similar weight loss kinetics (Fig. 2a) and consequent survival kinetics (Fig. 2b) of infected mice compared to infected control mice administered compressed room air.
Continuous or intermittent iNO administration does not reduce lung viral load
As gaseous NO (gNO) at high concentrations has been shown to decrease the viral load of infected cells in vitro (Miller C, personal communication), we examined whether iNO could reduce the viral load of influenza virus-infected mice. iNO was administered starting 1 hour prior to influenza WSN/33 infection and continued either continuously at 80 ppm or intermittently at 160 ppm for 30 min every 3.5 hours until mouse lungs were harvested at peak influenza viral load in the lungs (determined to be day 5 post-infection based on preliminary studies, data not shown). Since iNO was administered both prior to and for 5 days post-infection, we were able to test whether iNO at intermediate (80 ppm) or high concentration (160 ppm) could prevent either viral entry or viral replication in vivo, and thereby reduce viral load. Continuous iNO at 80 ppm, intermittent iNO at 160 ppm, and compressed room air administration yielded similar lung viral loads of infected mice on day 5 post-infection (Fig. 3a and b, respectively). Therefore, both continuous and intermittent iNO administration failed to reduce lung viral load of infected mice, compared to infected control mice administered compressed room air.
Figure 1
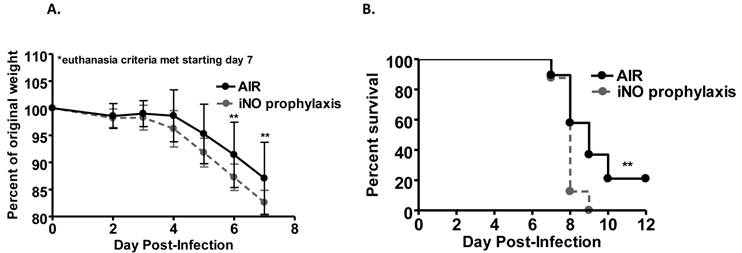
Prophylactic iNO therapy increased weight loss and decreased survival of C57Bl/6 mice infected with influenza A/WSN/33. C57Bl/6 male mice were infected with 103 PFU WSN/33 and administered continuous NO at 80 ppm (grey) or compressed room air (black) starting 1 hour prior to infection (n=17-18/group, two independent pooled experiments). (a) Mice receiving iNO displayed a significant reduction in weight compared to infected controls (Two-way ANOVA p < 0.001 with Bonferroni post-tests: P < 0.01 on day 6 and 7 post-infection). Error bars represent standard deviations. (b) iNO significantly reduced survival of treated mice compared to infected controls as shown by Kaplan-Meir survival curves (log-rank test: P < 0.01).
Figure 2
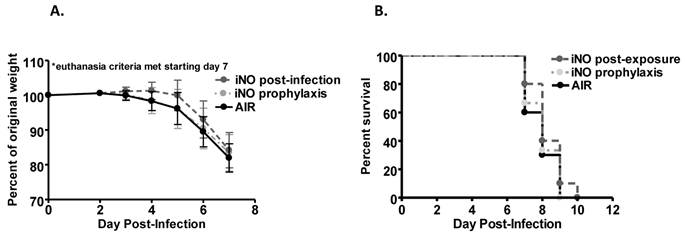
Prophylactic and post-infection intermittent iNO did not alter (a) weight loss kinetics or (b) survival of C57Bl/6 mice infected with 103 PFU WSN/33. C57Bl/6 male mice were infected with 103 PFU of WSN/33 and administered intermittent NO at 160 ppm for 30 min intervals every 3.5 hours starting either 1 hour prior to infection (light grey) or 4 hours post-infection (dark grey). Infected controls were administered compressed room air (black) (n=9-10/group). Error bars represent standard deviations.
Figure 3
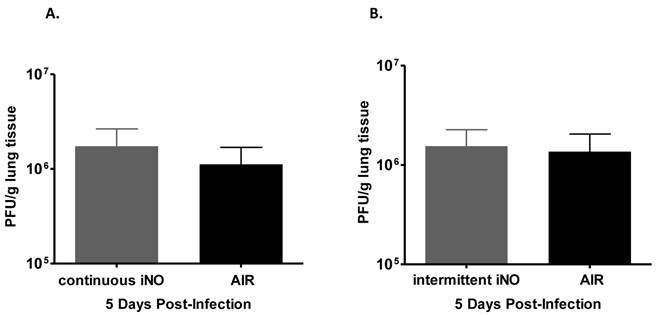
Intermittent high dose iNO prophylactic therapy failed to decrease viral load of C57Bl/6 mice infected with influenza WSN/33. Lungs were collected 5 days post-WSN/33 infection from mice treated with (a) continuous NO at 80 ppm (grey) or compressed room air (black) starting 1 hour prior to infection (n=5/group) or (b) intermittent NO at 160 ppm (grey) or compressed room air (black) for 30 min intervals every 3.5 hours starting 1 hour prior to infection (n=5/group). Error bars represent standard deviations. Lung viral load was quantified for all experimental groups by plaque assay on MDCK cells.
DISCUSSION
iNO therapy is currently FDA approved for the treatment of term and near-term neonates with hypoxemic respiratory failure associated with clinical or echocardiographic evidence of pulmonary arterial hypertension (26,27). Variable findings have been reported for iNO efficacy when administered at 1 ppm and up to 80 ppm. For its indicated use, iNO has been found to increase vasodilation, improve oxygenation, reduce length of mechanical ventilation, reduce oxygen requirement, and decrease length of stay in the intensive care unit (27, 30, 31). However, systematic reviews have failed to demonstrate that iNO therapy reduces overall mortality (32, 33).
Systematic reviews and meta-analysis of randomized controlled trials have shown that iNO, when used therapeutically in the management of ARDS, results in a transient improvement in arterial oxygenation but does not reduce mortality (34-36). Moreover, iNO therapy for ARDS may increase the risk of iNO treated patients developing renal dysfunction (35, 36). Despite this, 39% of critical care specialists surveyed reported using iNO for the management of patients with ARDS in Ontario, Canada (37).
Typically, iNO is administered at initial doses of 5-20 ppm in randomized controlled trials and observational studies for neonatal hypoxic respiratory failure (27). Although FDA-approved at concentrations up to 80 ppm, no specific dose of iNO has been proven more advantageous than another (27, 34). Rather, methemoglobinemia, defined as 7% methemoglobin in Davidson et al. (38), was more likely to occur. Methemoglobinemia may account for the decrease in survival observed in our study with continuous iNO administration at 80 ppm. NO2 concentrations were measured daily over the course of infection and kept below 2 ppm as is acceptable in humans, however, lung toxicity may still explain these results as the toxic threshold in mice may be lower. On the other hand, given previous in vitro findings by McMullen et al. (28), 80 ppm may also have been too low of a concentration to provide an antiviral effect.
A high dose of NO at 160 ppm was administered intermittently, not to target the airway vessels specifically, but rather to induce an antimicrobial effect while avoiding the harmful effects of high dose continuous iNO delivery. iNO administered to influenza infected mice in this manner, either prophylactically or therapeutically, failed to improve survival of infected mice, change the course of weight loss, or decrease the lung viral load, compared to control mice receiving compressed air. Therefore, although administration of high dose intermittent iNO may have reduced the harmful side-effects of NO, antimicrobial activity was not observed in vivo.
In conclusion, despite the demonstrated antimicrobial activity of NO against influenza A virus in vitro, the results of this study do not support the use of iNO as a prophylactic or treatment strategy to reduce viral burden or improve clinical outcome in severe influenza in vivo. Furthermore, it may be difficult to achieve viricidal concentrations of NO in the airways using iNO at concentrations that are safe in the living host.
MATERIALS AND METHODS
Murine influenza model
Animal use protocols were reviewed and approved by the University Health Network Ontario Cancer Institute Animal Care Committee, and all experiments were conducted in accordance with institutional guidelines in an animal biosafety level 2 facility. Female C57Bl/6 mice, 9-11 weeks old, were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and maintained under pathogen-free conditions with a 12-hour light cycle. On day 0, while under light isofluorane anesthesia, experimental mice were infected via nasal instillation with 103 plaque forming units (PFU) of influenza A/WSN/33 (H1N1; WSN/33,) (stock kindly provided by Dr. Eleanor Fish, University Health Network/University of Toronto) in 50 μl PBS. Weight was recorded daily for a maximum of twelve days post-infection, and mice were sacrificed when euthanasia criteria was met (greater than 20% weight loss). Lung tissue was harvested for analysis on day 5 post-infection.
In Vivo NO Delivery
Prophylactic or post-infection iNO therapy was initiated either 1 hour prior to or 4 hours post-infection, respectively. Mice were placed in flow-through chambers with free access to food and water and received either compressed room air, continuous NO at 80ppm +/-5ppm mixed with compressed room air, or intermittent NO for 30 min every 3.5 hours at 160ppm+/-5ppm mixed with compressed room air. Soda lime (200 g) was supplied to each chamber, and gas flow was maintained at 10-12 L/min to scavenge and minimize NO2 levels, respectively. NO2 levels were limited to <2 ppm for continuous iNO therapy and <8 ppm for intermittent iNO therapy. NO and NO2 levels were measured using an AeroNOX machine (Pulmonox Medical, AB, CA).
Lung influenza viral load analysis
Lungs were harvested and frozen at -80°C. Lungs were thawed, weighed, and homogenized in 1 ml PBS for 30 sec using a Tissue Miser homogenizer (Fisher Scientific, ON, CA). Lung homogenates were spun at 10,000xg for 10 min, aliquoted, and stored at -80°C for viral yield titration. Influenza WSN/33 viral yield in lung homogenates was quantified by plaque assay in MDCK canine kidney epithelial cells (ATCC, VA, USA). Cells were maintained in Eagle's MEM (ATCC, VA, USA) supplemented with 10% fetal bovine serum and antibiotics. MDCK cells were cultured at 37°C with 5% CO2. Cells were plated at a concentration of 8x106 cells/plate in 6 well culture plates. 12-24 hours later, medium was removed and MDCK cells were washed twice with PBS. 10-fold dilutions of lung homogenates were added to MDCK cells in 500 μL Eagle's MEM, in duplicate, and incubated at 37°C with 5% CO2 for 1 hour with plates rocked every 15 min. After incubation, 1 mL of serum-free 2X Eagle's MEM supplemented with 8 μl/ml trypsin, 60 μl/ml of 7.5% sodium bicarbonate and 20 μl/ml antibiotics, combined with 1 ml of 1.2% agarose, was added to each well. Once the agarose set, plates were incubated at 37°C for 42-72 hours until syncitia were observed. Plates were fixed with Carnoy's fixative (3:1, methanol:glacial acetic acid) for 30 min then stained with 0.1% crystal violet in 20% ethanol to visualize plaques. Viral load is expressed as plaque forming units per gram of lung tissue (PFU/g).
Statistical Analysis
Log-rank tests were performed on Kaplan-Meier survival curves. Significant differences in weight loss between groups were assessed by two-way analysis of variance (ANOVA) and Bonferroni post-tests were performed. A Student's t-test was carried out on viral yield data to assess significant differences (p ≤ 0.05) between experimental groups.
Acknowledgements
This work was supported by a Canadian Institutes of Health Research (CIHR) Masters Award [ID], a Defence Advanced Research Projects Agency Grant DARPA-BAA-09-43 [WCL, KCK, CM], and Canada Research Chairs (WCL, KCK) from the Canadian Institutes of Health Research (CIHR) (MOP-244701 and 13721 [KCK]). The authors sincerely thank Ms. Beata Majchrzak, Dr. Eleanor Fish, Dr. Hani Kim, and Dr. Lena Serghides, for their helpful advice and insights on this project.
CONFLICT OF INTEREST
CM consults and has minority shares in a variety of companies developing unrelated nitric oxide products. ID, KCK, and WCL have declared that no conflict of interest exists.
References
1. WHO: Geneva, Switzerland. Fact sheet 211, Influenza. http://www.who.int/mediacentre/factsheets/fs211/en/
2. Sheu TG, Deyde VM, Okomo-Adhiambo M. et al. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob Agents Chemother. 2008;52:3284-92
3. 2008-2009 Influenza season week 39 ending October 3, 2009. CDC. http://www.cdc.gov/flu/weekly/weeklyarchives2008-2009/weekly39.htm
4. Influenza A (H1N1) virus resistance to oseltamivir. Geneva, Switzerland: WHO. http://www.who.int/influenza/resources/documents/H1N1webupdate20090318_ed_ns.pdf
5. Weekly update on oseltamivir resistance to influenza H1N1 (2009) viruses. Geneva, Switzerland: WHO. http://www.who.int/influenza/2011_02_11_weekly_web_update_oseltamivir_resistance.pdf
6. WHO: Geneva, Switzerland. Weekly epidemiological record. 2011; 86: 81-92. http://www.who.int/wer/2011/wer8610.pdf
7. Le QM, Kiso M, Someya K. et al. Avian flu: Isolation of drug-resistant H5N1 virus. Nature. 2005;437:1108
8. De Jong MD, Thanh TT, Khanh TH. et al. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N Engl J Med. 2005;353:2667-72
9. Sheu TG, Fry AM, Garten RJ. et al. Dual resistance to adamantanes and oseltamivir among seasonal influenza A (H1N1) viruses: 2008-2010. J Infect Dis. 2011;203:13-17
10. Murphy BR, Baron S, Chalhub EG, Uhlendorf CP, Chanock RM. Temperature-sensitive mutants of influenza virus. IV. Induction of interferon in the nasopharynx by wild-type and a temperature- sensitive recombinant virus. J Infect Dis. 1973;128:488-93
11. Aoki FY, Macleod MD, Paggiaro P. et al. Early administration of oral oseltamivir increases the benefits of influenza treatment. J Antimicrob Chemother. 2003;51:123-29
12. Kandun IN, Tresnaningsih E, Purba WH. et al. Factors associated with case fatality of human H5N1 virus infections in Indonesia: A case series. Lancet. 2008;372:744-49
13. Jain S, Kamimoto L, Bramley AM. et al. Hospitalized patients with 2009 H1N1 influenza in the United States, April-June 2009. N Engl J Med. 2009;361:1935-44
14. To K, Hung I, Li I. et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin Infect Dis. 2010;50:850-59
15. Kumar A. Early versus late oseltamivir treatment in severly ill patients with 2009 pandemic influenza A (H1N1): speed is life. J Antimicrob Chemother. 2011;66:959-63
16. Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907-16
17. Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies (Review). Nat Rev Microbiol. 2004;2:820-32
18. Fang FC, De Groote MA. NO inhibitions: antimicrobial properties of nitric oxide. Clin Infect Dis. 1995;21:S162-5
19. Akerstrom S, Mousavi-Jazi M, Klingstrom J, Leijon M, Lundkvist A, Mirazimi A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J Virol. 2005;79:1966-69
20. Rimmelzwaan G, Baars M, Lijster P, Fouchier R, Osterhaus A. Inhibition of influenza virus replication by nitric oxide. J Virol. 1999;73:8880-3
21. Karupiah G, Xie QW, Buller RML. et al. Inhibition of viral replication by IFNg induced NO synthase. Science. 1993;261:1445-8
22. Croen KD. Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J Clin Invest. 1993;91:2446-52
23. Chen L, Liu P, Gao H. et al. Inhalation of nitric oxide in the treatment of severe acute respiratory syndrome: a rescue trial in Beijing. Clin Infect Dis. 2004;39:153135
24. Napolitano LM, Park PK, Raghavendran K, Bartlett RH. Nonventilatory strategies for patients with life-threatening 2009 H1N1 influenza and severe respiratory failure. Crit Care Med. 2010;38:e74-90
25. Kumar A, Zarychanski R, Pinto R. et al. Critically ill patients with 2009 influenza A (H1N1) infection in Canada. JAMA. 2009;302:1872-79
26. Food and Drug Administration. Approval of NDA 20-846 INOmax nitric oxide gas. USA: FDA. 1999
27. DiBlasi RM, Myers TR, Hess DR. Evidence-based clinical practice guideline: inhaled nitric oxide for neonates with acute hypoxic respiratory failure. Respir Care. 2010;55:1717-45
28. McMullin B, Chittock D, Roscoe D, Garcha H, Wang L, Miller C. The antimicrobial effect of nitric oxide on the bacteria that cause nosocomial pneumonia in mechanically ventilated patients in the ICU. Resp Care. 2005;50:1451-56
29. Miller C, McMullin B, Ghaffari A, Miller J, Stenzler A, Pick N, Roscoe D, Ghahary A, Road J, Av Gay Y. Gaseous nitric oxide bactericidal activity retained during intermittent high-dose short duration exposure. Nitric Oxide. 2009;20:16-23
30. Clark RH, Kueser TJ, Walker MW. et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical inhaled nitric oxide research group. N Engl J Med. 2000;342:469-74
31. The Franco-Belgium Collaborative NO Trial Group. Early compared with delayed inhaled nitric oxide in moderately hypoxaemic neonates with respiratory failure: a randomised controlled trial. Lancet. 1999;354:1066-71
32. Barrington KJ, Finer N. Inhaled nitric oxide for respiratory failure in preterm infants. Cochrane Database Syst Rev. 2010;12:CD000509
33. Donohue PK, Gilmore MM, Cristofalo E. et al. Inhaled nitric oxide in preterm infants: a systematic review. Pediatrics. 2011;127:e414-22
34. Sokol J, Jacobs SE, Bohn D. Inhaled nitric oxide for acute hypoxemic respiratory failure in children and adults: a meta-analysis. Anesth Analg. 2003;1:CD002787
35. Adhikari NKJ, Burns KEA, Friedrich JO, Granton JT, Cook DJ, Meade MO. Effect of nitric oxide on oxygenation and mortality in acute lung injury: systematic review and meta-analysis. BMJ. 2007;334:779
36. Afshari A, Brok J, Møller AM, Wetterslev J. Inhaled nitric oxide for acute respiratory distress syndrome and acute lung injury in adults and children: a systematic review with meta-analysis and trial sequential analysis. Anesth Analg. 2011;112:1411-21
37. Meade MO, Jacka MJ, Cook DJ. et al. Survey of interventions for the prevention and treatment of acute respiratory distress syndrome. Crit Care Med. 2004;32:946-54
38. Davidson D, Barefield ES, Kattwinkel J. et al. Inhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: a randomized, double-masked, placebo-controlled, dose-response, multi-center study. The I-NO/PPHN Study Group. Pediatrics. 1998;1:325-34
Author contact
![]() Corresponding author: conrad.lilesca
Corresponding author: conrad.lilesca
Citation styles
APA
Darwish, I., Miller, C., Kain, K.C., Liles, W.C. (2012). Inhaled Nitric Oxide Therapy Fails to Improve Outcome in Experimental Severe Influenza. International Journal of Medical Sciences, 9(2), 157-162. https://doi.org/10.7150/ijms.3880.
ACS
Darwish, I.; Miller, C.; Kain, K.C.; Liles, W.C. Inhaled Nitric Oxide Therapy Fails to Improve Outcome in Experimental Severe Influenza. Int. J. Med. Sci. 2012, 9 (2), 157-162. DOI: 10.7150/ijms.3880.
NLM
Darwish I, Miller C, Kain KC, Liles WC. Inhaled Nitric Oxide Therapy Fails to Improve Outcome in Experimental Severe Influenza. Int J Med Sci 2012; 9(2):157-162. doi:10.7150/ijms.3880. https://www.medsci.org/v09p0157.htm
CSE
Darwish I, Miller C, Kain KC, Liles WC. 2012. Inhaled Nitric Oxide Therapy Fails to Improve Outcome in Experimental Severe Influenza. Int J Med Sci. 9(2):157-162.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.