Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
INTRODUCTION
CLASSIFICATION
CLINICAL PRESENTATION
EPIDEMIOLOGY
STARVATION MODEL
CRF MECHANISM
OPIOID PEPTIDES
GHRELIN
LEPTIN
NEURO-PERIPHERY MECHANISMS OF...
SEX DIFFERENCES IN THE CRF,...
DISCUSSION
Acknowledgements
Supplementary Material
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2011; 8(8):679-703. doi:10.7150/ijms.8.679 This issue Cite
Review
Anorexia Nervosa: A Unified Neurological Perspective
Tasneem Fatema Hasan ![]() , Hunaid Hasan
, Hunaid Hasan ![]()
Mahatma Gandhi Mission's Medical College, Aurangabad, Maharashtra, India
Received 2011-5-3; Accepted 2011-9-19; Published 2011-10-22
Abstract
The roles of corticotrophin-releasing factor (CRF), opioid peptides, leptin and ghrelin in anorexia nervosa (AN) were discussed in this paper. CRF is the key mediator of the hypothalamo-pituitary-adrenal (HPA) axis and also acts at various other parts of the brain, such as the limbic system and the peripheral nervous system. CRF action is mediated through the CRF1 and CRF2 receptors, with both HPA axis-dependent and HPA axis-independent actions, where the latter shows nil involvement of the autonomic nervous system. CRF1 receptors mediate both the HPA axis-dependent and independent pathways through CRF, while the CRF2 receptors exclusively mediate the HPA axis-independent pathways through urocortin. Opioid peptides are involved in the adaptation and regulation of energy intake and utilization through reward-related behavior. Opioids play a role in the addictive component of AN, as described by the “auto-addiction opioids theory”. Their interactions have demonstrated the psychological aspect of AN and have shown to prevent the functioning of the physiological homeostasis. Important opioids involved are β-lipotropin, β-endorphin and dynorphin, which interact with both µ and κ opioids receptors to regulate reward-mediated behavior and describe the higher incidence of AN seen in females. Moreover, ghrelin is known as the “hunger” hormone and helps stimulate growth hormone (GH) and hepatic insulin-like-growth-factor-1(IGF-1), maintaining anabolism and preserving a lean body mass. In AN, high levels of GH due to GH resistance along with low levels of IGF-1 are observed. Leptin plays a role in suppressing appetite through the inhibition of neuropeptide Y gene. Moreover, the CRF, opioid, leptin and ghrelin mechanisms operate collectively at the HPA axis and express the physiological and psychological components of AN. Fear conditioning is an intricate learning process occurring at the level of the hippocampus, amygdala, lateral septum and the dorsal raphe by involving three distinct pathways, the HPA axis-independent pathway, hypercortisolemia and ghrelin. Opioids mediate CRF through noradrenergic stimulation in association with the locus coeruleus. Furthermore, CRF's inhibitory effect on gonadotropin releasing hormone can be further explained by the direct relationship seen between CRF and opioids. Low levels of gonadotropin have been demonstrated in AN where only estrogen has shown to mediate energy intake. In addition, estrogen is involved in regulating µ receptor concentrations, but in turn both CRF and opioids regulate estrogen. Moreover, opioids and leptin are both an effect of AN, while many studies have demonstrated a causal relationship between CRF and anorexic behavior. Moreover, leptin, estrogen and ghrelin play a role as predictors of survival in starvation. Since both leptin and estrogen are associated with higher levels of bone marrow fat they represent a longer survival than those who favor the ghrelin pathway. Future studies should consider cohort studies involving prepubertal males and females with high CRF. This would help prevent the extrapolation of results from studies on mice and draw more meaningful conclusions in humans. Studies should also consider these mechanisms in post-AN patients, as well as look into what predisposes certain individuals to develop AN. Finally, due to its complex pathogenesis the treatment of AN should focus on both the pharmacological and behavioral perspectives.
Keywords: anorexia nervosa, corticotrophin releasing factor, opioids peptide, ghrelin, leptin, sex differences, energy balance
INTRODUCTION
Anorexia Nervosa (AN) is an eating disorder that continues to show devastating effects on both adolescents and adults, worldwide. The Diagnostic and Statistical Manual of Mental Disorders has defined AN as an eating disorder in which people refuse to maintain a minimally required healthy weight for their age and height (body weight less than eighty five percent of expected), have an intense fear of gaining weight and significantly misinterpret their body and shape (1).
CLASSIFICATION
Kaye WH (1996) for academic purposes has classified patients into three different categories, low weight, short-term recovered and long-term recovered (2). The American Psychiatric Association (1994) has subdivided AN into two distinct categories. The first category is the restrictive type (RAN), where patients exhibit “restricted food intake without binge eating or purging,” while the second category is the binge-eating/purging type (BPAN), involving both “binge eating/purging episodes during anorexia and bulimia phases” (3). In addition, both categories can also be differentiated by their hormonal profiles, such that lower leptin levels have been found in RAN (4). On the other hand, increased impulsivity and higher rate of self-harm have been observed in BPAN (4-6).
CLINICAL PRESENTATION
There are repeated acts of “body checking” where anorexics constantly and obsessively monitor their body image to assure that they are thin (7). Clinically, AN is differentiated on the basis of RAN and BPAN. In RAN, patients experience weight loss by significantly reducing their total calorie intake along with exaggerated physical work-outs. In BPAN, patients resort to vomiting and the use of laxatives or diuretics to stay thin (1).
Moreover, the clinical features of AN can be further divided as mental and physical. In general, anorexics secretly use aberrant, unusual behavior to lose weight. They gradually refuse eating with family and out in public (1). Although, a loss of appetite is seen late in the course of the disorder, collecting recipes and preparing fancy meals for others is evidence that the individual is constantly thinking of food (1). Moreover, carrying large amounts of candy in pockets, hiding food throughout the house, disposing food in napkins and cutting meals into small pieces and rearranging them on the plate are important details that give insight into the character of this disorder (1). In addition, compulsive stealing of candy and laxatives are also seen (1). Anorexics find opportunities to stay physically active, ranging from athletics and dance to acts as simple as, preferring to stand rather than sit (7). Nevertheless, they show resistance when offered help, and refuse to talk when confronted about their unusual behavior (1). Although, perfectionists by nature, anorexics are socially-isolated individuals that also frequently suffer from depression, anxiety and obsessive-compulsive disorder (OCD) (1). Suicidal tendencies are commonly encountered in patients suffering from BPAN (1). A mental status examination reveals that the individual is alert, oriented and knowledgeable on the topic of nutrition (1).
The profound weight loss observed in AN results in hypothermia (body temperature of about 35 degrees Celsius), hypotension, dependent edema, bradycardia, lanugo and various metabolic changes (1). Changes like amenorrhea, poor sexual adjustment and epigastric complaints are also commonly observed (1). In BPAN, patients suffer from hypokalemic alkalosis due to self-induced vomiting and the abuse of purgatives (1). Moreover, an electrocardiogram reflects a flattening and inversion of T waves, depression of ST segment and lengthening of QT interval in the emaciated stage (1). Rare complications like gastric dilation and superior mesenteric artery syndrome have also been noted (1).
EPIDEMIOLOGY
AN is common between the ages of ten to thirty years, with the greatest incidence seen at seventeen to eighteen years of age (1). The prevalence of AN is between 0.3-0.6% (8,9) with a mortality rate of 5-18% per decade, possibly due to cachexia and suicide (1,10,11). A survival analysis performed by Carter JC et al. (2004) demonstrated an overall relapse rate of 35% and mean survival time of eighteen months (12). The high risk period for relapse was defined between six to seventeen months after discharge (12). Key predictors for relapse were history of previous treatment, history of suicide attempt, associated OCD symptoms at presentation, concern for body image at discharge and initiation of excessive physical activity after discharge (12). Moreover, Talbot Y (1983) reported a cure rate of 70% (13). Recent sources have suggested that 75-85% of anorexics are likely to fully recover (14). This estimate would increase to 90% if patients undergoing profound recovery were to be included (14). In addition, AN is a female dominant disorder. For every one to three males, nine females have shown to suffer from AN (8,9).
Traditionally, AN was a disorder giving equal emphasis on the biological, psychological and sociological dysfunction. However, recent evidence has found a higher predilection towards the biological perspective, shifting from the bio-psycho-social model to the biological model. Twin studies have suggested a moderate to high heritability (50-85%) of AN (15-19). In support, an adoption study performed by Klump KL et al. (2009) found similar findings (59-82%) (16), while other studies also showed heritability of 70% (9,20).
Furthermore, research on AN has looked into the dysfunction of various neuropeptides at the level of the central nervous system (CNS) and the peripheral nervous system (PNS). Therefore, this paper will review the evidence supporting the implications of corticotrophin-releasing factor (CRF), opioid peptides, ghrelin and leptin in the pathogenesis of AN. Uniquely, this paper will take an integrative approach to bring this evidence together to propose a more holistic and complete model for AN.
STARVATION MODEL
It is essential to understand the physiology of starvation to better understand AN. Many effects of AN are regulated through the starvation response. The starvation response consists of three phases (21). Phase one is the period when the consumed meal has been digested and the body has entered the post-absorptive phase (22). The first phase is brief and usually does not store any glucose or glycogen for energy (22). The total body glucose and glycogen stores are four hundred eighty grams, which are usually exhausted within twenty-four hours (23). Phase two becomes prominent when glycogen stores completely deplete. A greater mobilization of fat is seen during this stage. This stage is responsible for many of the physiological and biochemical alterations in the body (22). Increase in free fatty acids (FFA) lead to an increase in the peroxisome proliferator-activated receptor (PPAR)-α and PPAR- ɣ (24,25). Next, PPAR-α increases levels of fibroblast growth factor-21. Fibroblast growth factor-21 mediates growth hormone (GH) resistance and reduces (insulin-growth-factor-1) IGF-1 levels (26,27). Further, if starvation continues, the fat stores exhaust and the body enters phase three of starvation. During this phase, there is a breakdown of muscle tissue and the amino acids liberated are used in the formation of glucose for maintaining brain function. This is called “protein wasting” (21,28). Therefore, adapting to starvation involves reducing energy expenditure by suppressing metabolic rate, body temperature and delaying growth/reproduction (29-31).
CRF MECHANISM
CRF is a 41-amino acid hypothalamic peptide vital for regulating adrenocorticotrophic hormone (ACTH) secretion (32-34) and neuroendocrine and behavioral stress-related responses (35-39). Numerous studies have demonstrated many visceral and behavioral effects of CRF (40). CRF has shown to activate the hypothalamo-pituitary-adrenal (HPA) axis and other various parts of the brain, specifically, the limbic system (34,37,38,41). Autoradiography has identified CRF receptors in the CNS and the PNS demonstrating various physiological actions of CRF (40).
Central and Peripheral Effects of CRF
CRF is governed by two groups of receptors, CRF1 and CRF2, belonging to the seven transmembrane family of receptors (42-44). The CRF1 receptors are found in the cerebral cortex and the anterior lobe of the pituitary gland mediating anxiogenic effects of CRF (45-54), while the CRF2 receptors have been found in the ventromedial hypothalamus (VMH) and the paraventricular nucleus of the hypothalamus (PVNH) (34). In addition, CRF-containing cell bodies have been identified in the PVNH with projections to the median eminence (55,56). Moreover, three splice variants of the CRF2 receptor, α, β and ɣ, have been recognized (57-60). The CRF2-α receptor, abundantly expressed in the hypothalamus and the limbic system, mediates anxiety, depression and feeding behavior (61), while CRF2-α protein and mitochondrial ribonucleic acid (mRNA), densely present in the septum, regulates emotional responses of fear, anxiety and aggression (62,63).
Moreover, CRF is also present in the nucleus accumbens, lateral hypothalamus, parabrachial nucleus and dorsal motor nucleus of the vagus, regulating control pathways for nutrient intake, independent of pituitary control (55,64,65). In addition, CRF is also found in those areas of the limbic system that control the central autonomic function (56,66-68).
The amygdala is responsible for causing reactions of arousal, attention, fear and rage associated with sympathetic nervous system (SNS) activation (69). Similar reactions have been observed after administrating CRF intracerebroventricularly (32). Therefore, the amygdala and the presence of CRF receptors are an important topic of discussion in AN. CRF receptors are densely located along the pathways to the frontal, orbital, cingulated, temporal and insular cortices (40). Moreover, the connections between the amygdala and the cortices are both afferent and efferent. Afferent connections are from the locus coeruleus, hypothalamus and dorsomedial thalamic nucleus, while the efferent connections are from the dorsomedial thalamic nucleus, nucleus stria terminalis, preoptic area, septal regions and the arcuate nucleus of the hypothalamus (ARCH), all of which contain CRF receptors (40). The other areas of the limbic lobe, such as, the cingulated, parahippocampal cortex and the hippocampus, all contain high concentrations of CRF receptors and are closely related to the hypothalamus and the neocortex (70).
The CRF receptors of rats and monkeys are found in the preoptic area and the ARCH, and have shown to regulate gonadotropin secretion (40). When CRF was injected into the ARCH and VMH of female rats, there was a decline in luteinizing hormone levels and inhibition of sexual behavior, suggesting that CRF mediates sexual behavior (71). Similarly, these observations were also seen in humans during conditions of prolonged stress (40).
Claes SJ (2004) suggested, “corticotrophin releasing hormone (CRH) is the most important hypothalamic peptide that controls HPA axis activity” (72). Intracerebroventricular administration of CRF to rats, dogs and monkey's activated both the HPA axis and the SNS, with visceral, metabolic (32,33,73) and behavioral changes (32,74). A study on CRH gene knockout demonstrated the impairment of the HPA axis function in mice (75).
Moreover, the locus coeruleus is connected to the hypothalamus, hippocampus, cerebral cortex, olfactory bulb, cerebellum and the spinal cord (40). Valentino RJ et al. (1983) noted the activation of local neurons when CRF was injected into the locus coeruleus of rats (76). Therefore, the locus coeruleus plays an “integrative role” within the CNS, endocrine system, autonomic system and the behavioral system due to its various connections and the presence of immunoreactive CRF and CRF receptors (40). This also suggests that CRF plays the role of a common neural transmission mediator (40).
Immunoreactive CRF and its receptors have been identified in peripheral tissues like the adrenal medulla and have shown to regulate the SNS (77-79). Activation of these CRF receptors affects the secretory activity of adrenal glands (40). Udelsman R et al. (1986) demonstrated that incubating CRF-containing cells for 24 hours resulted in dose-dependent stimulation of epinephrine, noradrenaline (NA) and met-enkephalin (80).
OPIOID PEPTIDES
Opioid peptides are responsible for adaptation and regulation of energy intake and utilization through reward-mediated behavior (81). They are the key mediators of hedonic balance and emotional response in food choice and intake (82).The opioid peptides, β-lipotropin (β-LP) and β β-endorphin (β-EP) are pro-opiomelanocortin (POMC)-derived and help regulate reward-mediated behavior (83,84). Another opioid peptide, dynorphin, a precursor of protein prodynorphin (83,84), maintains homeostasis through appetite control and circadian rhythms (83).
Types of opioid peptides
Opioid peptides are categorized as δ, µ and κ. These opioids occupy the nucleus tractus solitarius, PVNH, VMH, amygdala, the perifornical area, nucleus accumbens and the forebrain regions (85-87). While µ and κ opioids regulate reward-mediated behavior, δ opioids are involved in self-stimulation (88).
GHRELIN
Ghrelin is an important gastrointestinal peptide hormone synthesized and secreted by the X/A-like cells in the oxyntic glands of the gastric fundic mucosa (89) and proximal small intestine (90). Ghrelin is an essential “hunger” hormone secreted during starvation (91). It regulates energy homeostasis by signaling the CNS to increase food intake and reduce energy expenditure (90,91). Ghrelin secretion occurs in a pulsatile manner, starting with a gradual pre-prandial rise, later peaking at meal initiation and finally reducing to baseline levels one hour after food intake (92-95). In sum, ghrelin secretes in response to reduced gastrointestinal content and returns to baseline levels upon food intake (92).
Ghrelin appears as a 117-amino acid pre-prohormone which breaks down post-translational into a 28-amino acid peptide (96) and acylates at its serine-3-residue by ghrelin O-acyl-transferase (GOAT) (97,98). Two forms of ghrelin have been identified, the active (acyl ghrelin) and inactive (des-acyl ghrelin) forms (99). When acyl ghrelin is released into the circulation, it lives a half-life of thirty-minutes and subsequently converts into its inactive form (99). Moreover, ghrelin presents as an endogenous ligand for the GH secretogogue-1a receptor in the hypothalamus and pituitary gland (90,100,101).
Ghrelin plays an essential role in feeding behavior. During meal initiation, ghrelin directly activates the medial ARCH neurons (102). Ghrelin-mediated stimulation of the hypothalamic GH secretogogue-1a receptor results in an anabolic response. This is evident by the exaggerated release of orexigenic peptides, neuropeptide Y (NPY) and agouti-related protein (AgRP), leading to an increase in food intake and decrease in energy expenditure (103-105).
Moreover, ghrelin stimulates the secretion of GH from the anterior pituitary gland with an indirect release of IGF-1 (90,106,107). Together, GH and IGF-1 help maintain lean body mass through anabolism (108-110). But in catabolic conditions like AN, GH encourages lipolysis and decreases glucose and protein oxidation in order to preserve lean body mass (109).
Injections of exogenous ghrelin have shown to increase the adiposity in rodents through its orexigenic effect (111-114). Similar findings are also demonstrated in humans through stimulation of appetite in the healthy and chronically-ill individuals (107,115,116). At pharmacological doses, ghrelin increases prolactin, ACTH and cortisol secretion (106). Lastly, episodes of food intake directly correlate with the levels of endogenous ghrelin (117) in both humans (94) and rats (118).
LEPTIN
Leptin is a four-helical structure consisting of 167-amino acids. It is analogous to a cytokine and is also known as a “prototypical adipokine” (119,120). Although, largely produced in the adipose tissue, leptin is expressed in various tissues like the placenta, ovary, mammary epithelium, bone marrow and lymphoid tissue (121,122).
Leptin secretion follows a circadian rhythm with the greatest secretion seen between midnight to early morning and lowest during early-to-mid-afternoon (123-125). Leptin has shown to suppress appetite by inhibiting NPY gene expression at the ARCH (126). Moreover, leptin concentrates directly correlate with the amount of adiposity in an individual, generally low during starvation and high in obesity (127). However, sudden changes in food intake, especially energy deprivation, results in wide fluctuation in the levels of leptin (128-130).
Moreover, females demonstrate greater plasma leptin concentrates than males, but these levels significantly decline after menopause (131). These differences are largely independent of body mass index (BMI), but can be attributed to differences in sex hormones, fat mass and body fat distribution (131-133). In addition, females tend to accumulate more peripheral amounts of body fat, while men exhibit an android distribution of fat. As a result, higher concentrates of leptin mRNA have been identified in the subcutaneous fat, but are scarcely present in the omentum (133,134). Therefore, this gives insight into why higher leptin levels are observed in females.
Leptin exerts its action through binding at the ObRs receptor in the CNS and at various peripheral tissues (135). Six isoforms of the ObRs receptor have been identified, ObRa, ObRb, ObRc, ObRd, ObRe and ObRf (136). Isoforms, ObRa and ObRc are vital in transporting leptin across the blood-brain barrier (137,138), while ObRb is primarily involved in leptin signaling (136,137,139). ObRb is chiefly demonstrable in the hypothalamus, regulating energy homeostasis and neuroendocrine function (135,140).
NEURO-PERIPHERY MECHANISMS OF ANOREXIA NERVOSA
CORTICOTROPHIN-RELEASING FACTOR
Physiological Perspective
Existing literature has attributed AN to the dysfunction of the CRF mechanism, with increased levels of CRH (141). Hotta et al. (1986) demonstrated high levels of CRF in the cerebrospinal fluid (CSF) of anorexics (308). Moreover, starvation has shown to activate the HPA axis (142-145). However, over-activity of the axis has been demonstrated by Carol BJ (1980) in depressed individuals (146). CRF receptors of the cerebral cortex and the limbic system manifest the visceral and behavioral components of depression (40). The clinical features attributed to CRF dysfunction and HPA-axis hyperactivity are: excessive physical activity, suppressed reproductive hormones resulting in decreased sexual behaviour and amenorrhea, cardiovascular changes like hypotension and bradycardia, anxiety, blunted social interaction, increased vigilance and altered immune system function (147-149). CRF has also shown to reduce food intake (39,149,150) and blunt weight gain (39,73,151-154), affecting both energy intake and utilization. However, Krahn et al. (1990) demonstrated that a persistent elevation of CRH was required to cause an AN-like syndrome, and an intermittent elevation had no effect (39). Anorexics possibly differ from healthy individuals in being unable to adapt to CRH elevations (39).
Smagin GN et al. (1998) found that CRF2 and not CRF1 antisense administration attenuated the effect of CRF on appetite (155). Urocortin (UCN), a CRH-related neuropeptide, demonstrates 20-40 times higher natural affinity to CRF2 receptors than CRF itself, resulting in suppression of appetite, independent of the HPA-axis and glucocorticoid release (156,157). Therefore, when dissociated from the pituitary, agonists to the CRF2 receptors have shown to suppress appetite, while the antagonists have shown to enhance appetite.
Cullen MJ et al. (2001) studied the effects of antisauvignine-30 (ASV-30), a CRF2-selective antagonist, on energy balance through the central infusion of CRF and UCN (44). Consequently, central infusion of CRF resulted in a negative energy balance attributed to decreased food intake and increased SNS activity. However, UCN only showed a minimum effect accounted by reduced food intake and nil involvement of the SNS. On the other hand, ASV-30 reversed the effects of both CRF and UCN by increasing food intake. However, ASV-30 failed to alter the effects of CRF on the HPA-axis variables like levels of corticosterone, increased adrenal weight, reduced thymus and splenic weight. Also, ASV-30 had a selective affect on CRF2 receptors, but demonstrated no metabolic effects of CRF (44). Moreover, Bornstein SR et al. (1998) suggested the role of CRH2 receptors in the anorexic effect of CRH through antalarmin administration, a CRH1 receptor antagonist (151). Finally, a series of other studies have demonstrated this relationship by performing an adrenalectomy in genetically obese animals. Results indicated that an increase in endogenous CRF in such animals resulted in reduced food intake and increased sympathetic activity (158-161).
Several studies have found a negative correlation between food intake and sympathetic activity (158,162,163). As a result of sympathetic innervation, brown adipose tissue (BAT) has the ability to undergo non-shivering thermogenesis, resulting in weight loss of CRF-infused rats. Sympathetic stimulation elevates norepinephrine, increases heart rate and releases glucocorticoids (164-171). Sympathetic stimulation-induced-lipolysis is supported by a rise seen in the levels of cholesterol, triglycerides and FFA in the circulation (158,162,163). More importantly, the sympathetic mechanism of the BAT functions independently of other bodily tissues (158,162,163).
Other studies have looked at CRF in the reverse relationship between food intake and energy utilization, mediated by the SNS (44). Arase K et al. (1988) explored the acute and chronic effects of CRF infusion in the third ventricle of rats (149). Acutely, CRF reduced food intake, but significantly increased sympathetic activity, while chronically, a prolonged but steady loss in weight was noted (149). Arase K et al. (1988) demonstrated that food intake and sympathetic stimulation were reciprocally-related when exploring the diurnal rhythm between both groups of rats (149). Moreover, rats under CRF-treatment demonstrated a low fat pad weight, suggesting that fat and muscle are possible sources of tissue loss under CRH-treatment. However, Cullen MJ et al. (2001) put forth that fat pad weight was an insensitive measure (44) and carcass fat is what was actually reduced after chronic central CRF infusion (172).
CRF in Conditioned Fear
An important component of AN is persistent fear. This fear is irrational and conditioned to weight gain. The model described for fear involves the formation of memory after an acute stressful event (173-175). The CRF released into the HPA-axis as a result of stress, further requires an interplay of several molecular processes (176) and hippocampal CRF receptor activation (173) for the formation of memory. As depicted in Figure 3, memory formation requires the interaction of two core signaling pathways, cyclic adenosine monophosphate -dependent protein kinases (PEK) and mitogen-activated extracellular signal-regulated kinases (Mek-1/2) (177-179). Elliott-Hunt CR et al. (2002) demonstrated that CRF helps in the activation of both PEK and Mek-1/2 through CRF1 and CRF2 receptors (180-182), and increased expression of CRF2 mRNA was shown to promote associative and stress-enhanced learning (176).
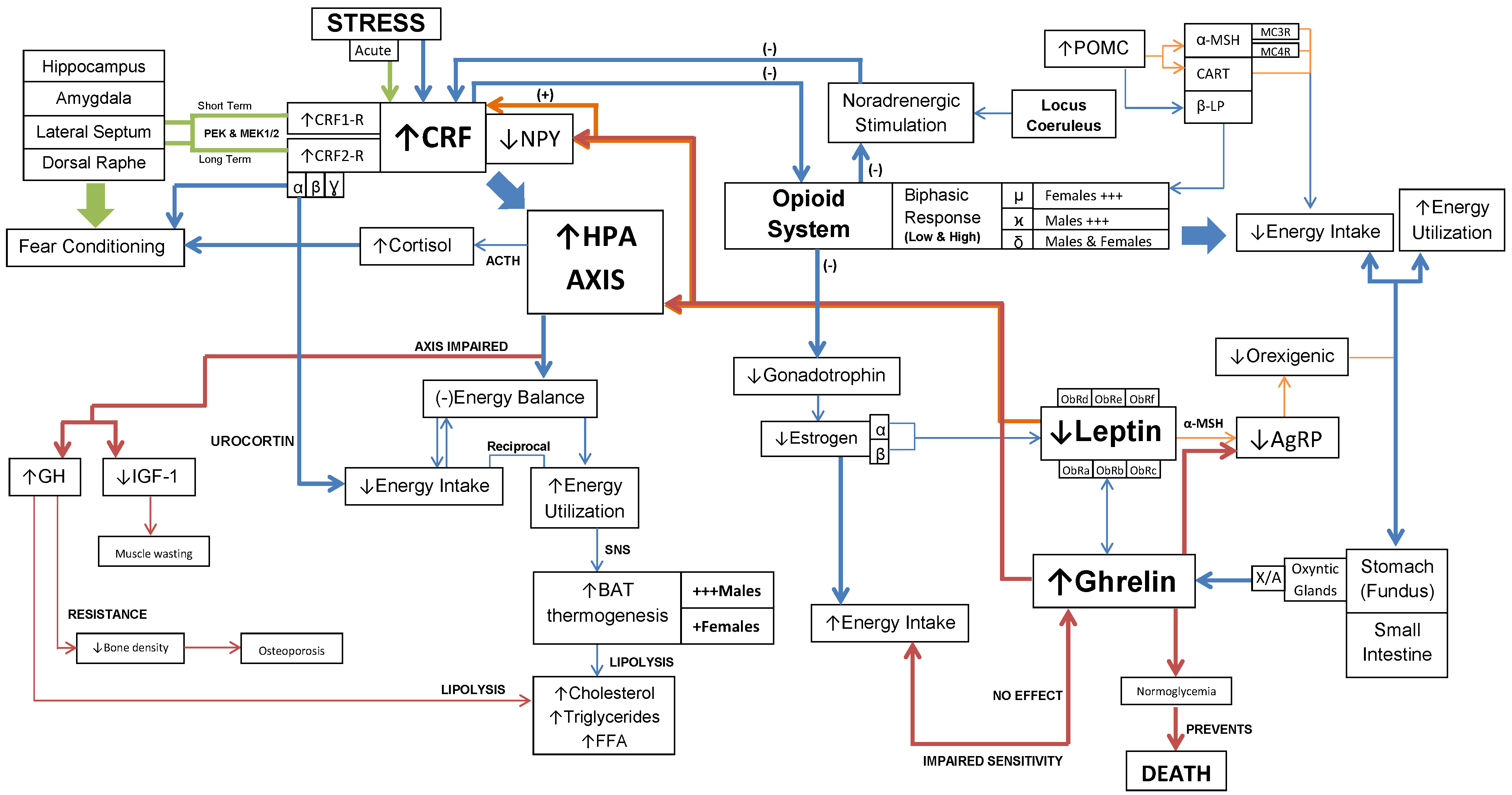
Interaction of CRF, opioids, ghrelin and leptin mechanisms in AN. This diagram represents the key pathways involved in the spectrum of physiological and psychological symptoms of AN.
Radulovic et al. (1999) found that injecting a nonselective CRF receptor antagonist, astressin, prevented the augmentation of fear conditioning (173). However according to Sananbenesi F et al. (2003), administering a selective CRF2 receptor antagonist, ASV-30, prevented fear conditioning after an acute stressful event (176).
Moreover, Ho et al. (2001) further evaluated the role of CRF2 receptors in fear conditioning by observing the shock-induced freezing response (61). Rats, treated with antisense oligonucleotides for 7 days, showed a 60-80% reduction in the overall effect of CRF2 receptors. Analgesic tests were used to control for loss of pain sensation. Therefore, inhibition of the CRF2 receptors in the lateral septum was shown to significantly reduce contextual fear conditioning (61). In addition, Hammack et al. (2003) also suggested that CRF2 receptors in the dorsal raphe were probably involved in the stress-mediated fear conditioning (183).
Nevertheless, it is important to consider the involvement of the amygdala in the formation of CRF-induced emotion arousing memories. The basolateral complex (BLA) and central nuclei of the amygdala have projection neurons with densely populated CRH receptors (184-186). Roozendaal B et al. (2002) found that injecting a CRF antagonist, α-helical CRF, in the BLA of the amygdala immediately post-training resulted in dose-dependent inhibitory avoidance retention impairment (175). Therefore, this suggests that the antagonist interfered with memory formation at the level of the BLA. In conclusion, the hippocampus, amygdala, lateral septum and dorsal raphe work collectively in the process of CRF-induced fear conditioning. Further, this sheds light upon the potential pharmacological interventions for treating fear complexes in AN.
Hypercortisolemia
Hypercortisolemia with elevated CRH is commonly seen in protein-caloric depleted anorexic patients (2). Hypercortisolemia is associated with excessive fear, atherosclerosis, osteoporosis and decreased immune function (72). Elevated cortisol has shown to suppress the mesolimbic-doparminogenic system (172), responsible for the reward-mediated behavior (187). Cortisol also regulates the negative feedback mechanism for CRH secretion. Possibly, the intense fear seen in AN can be explained by the rise in CRH and cortisol levels.
Psychological Perspective
Heinrichs et al. (1993) studied the mechanism of CRF-mediated feeding and proposed that NPY and CRF work collectively to regulate feeding behavior (188). CRF and endogenous NPY were found to work in opposite directions in modifying the behavioral and physiological effects of AN (189-192). Moreover, NPY has found to be most potent when injected nearby to the CRF neurons at the PVNH (193-196) and during HPA-axis activation (197-198). NPY has also shown to potentiate feeding through a negative glucocorticoid feedback mechanism and by a direct receptor antagonism at the PVNH (188). High levels of NPY and greater mRNA expression in the NPY neurons have been demonstrated in food deprived rats. However, these levels return to baseline upon re-feeding (189-201). Many behavioral studies have observed a psychological basis of how NPY invokes feeding behavior. It has been thought that NPY helps motivate eating. Therefore, dysfunctional NPY with CRF function influence the nature of feeding observed in AN, resulting in psychological alterations like, motivation towards dieting, psychosocial influences and stress (2).
Moreover, CRH production takes place at both the hypothalamus and the amygdala. CRH from the hypothalamus is reactive to the physiological aspects of AN, while that from the amygdala is reactive to psychological stress (72). Since AN consists of both a physiological and psychological component, this implies that CRH from both the hypothalamus and amygdala are responsible for anorexic behavior as a function of stress. Further, Kaye WH (1996) found a correlation between depression and CRH in those individuals that were psychologically dissatisfied with their weight, and not in subjects of constitutional thinness (SOCT) (2). In support, Pacak et al. (2002) also looked at depressed individuals with suicidal tendencies and demonstrated high levels of CRH in the locus coeruleus, median raphe and caudal dorsal raphe by 30%, 39% and 45%, respectively (172).
OPIOID PEPTIDES
Neurological Perspective
Opioids are responsible for regulating feeding behavior (81). Hubner HF (1993) found that administering naloxone (opioid antagonist) to anorexics resulted in weight gain, suggesting that opioids were potential mediators of anorexic behavior (202). A study by Abbate-Daga G et al. (2007) compared opiate-addicts to anorexic men and found similarities in the following personality traits: anxiety, fearfulness and antisocial features (203). However, there were distinct differences between both groups. Anorexic men displayed a higher persistence, but a low reward-dependence, while opiate-addicts were high novelty seekers and scored better on self-transcendence (203). Therefore, key differences in the pathogenesis of opiate-addiction and AN do clearly exist. Furthermore, an atypical endogenous opioid system seems to be present in anorexics, thus biologically predisposing them to develop AN (204). As discussed earlier, this supports the high heritability of AN, and suggests that the psychological component of AN is perhaps biologically-determined.
Lesem et al. (1991) observed that CSF levels of dynorphins were at normal values during all stages of AN (2). Moreover, opioids like β-EP are considered important in symptom perpetuation and relapses seen in AN. However, β-EP levels have shown to normalize after weight gain (202,205-207). Studies have also found a normal to reduced β-EP level in the CSF of anorexics (208). Hubner HF (1993) suggested that β-EP levels exhibit a biphasic effect on food and weight regulation (202). Therefore, both low and high levels of β-EP have shown to inhibit feeding (2,141). Kaye WH et al. (1987) further concluded that low levels of β-EP persist, but as patients recover, β-EP levels also normalize (206). Moreover, while low levels of plasma β-EP have been demonstrated in anorexics (84, 209), Tepper et al. (1992) found a significantly elevated level of β-EP in AN (210). In addition, Brambilla F et al. (1991) have demonstrated elevated levels of β-LP in anorexics (209).
To add to this phenomenon, Brambilla F et al. (1991) studied the dynamic peripheral secretion of β-EP and β-LP in AN (209). It was observed that both peptides were constantly elevated over a 24-hour period, particularly during the night (209). This suggests the involvement of the POMC system. However, a disassociation in the secretion of β-EP and β-LP was noticed, where β-EP was secreted only during the early hours of the night, and β-LP was secreted both during the day and at night (209). This implies that independent sources and regulatory methods for both peptides exist (209). Furthermore, studies have found an intermediate layer that exists in the human pituitary between the anterior and posterior lobes. This layer contains β-EP staining cells that have shown to increase during physiological and pathological conditions. Therefore, the disassociation between both peptides is possibly due to secretion from an alternate focus (209,211,212). In support, Brambilla F et al. (1991) concluded that the anterior pituitary POMC hypersecretion was due to starvation (209). However, β-EP has no such relation. Also, β-LP, not β-EP, was linked to weight loss, suggesting that β-EP secretes from an alternate focus (209). All in all, eating disorders which range from obesity to AN have three dysfunctional components affecting hunger and satiety: abnormal levels of peripheral β-EP and β-LP secretion, dysfunctional circadian rhythm and POMC peptide disassociation (209).
Brambilla F et al. (1991) also observed disruption in the normal rhythmicity of β-EP and β-LP secretion, while cortisol secretion continued to follow a normal pattern (209). This further supports the disassociation seen in the POMC-derived peptides, suggesting a disassociation of the hypothalamic and suprahypothalamic function (209).
Moreover, Glass et al. (2003) experimented with rats and provided evidence on the effect of different opioids on deprivation-induced feeding (213). When δ2-opioid antagonist, naltrindole isothiocyanate, was injected into the ventral tegmentum, deprivation-induced feeding showed insignificant changes. However, when κ antagonist, norbinaltorphimine was injected, deprivation-induced feeding significantly declined. Similarly, µ opioids demonstrated the most significant decline in food intake among all opioids (213). Since there was a profound reduction in food intake, this implies that AN is perhaps due to the malfunction of the facilitation of reward system mediated by the antagonistic κ and µ opioids (213). Moreover, the CSF of wasted anorexics has shown high levels of those substrates that are mediated through the µ receptors (207). Certain antagonistic δ receptors have also shown a statistically significant effect on reduced food intake. Moreover, self-stimulation plays a role in regulating anorexic behavior. An opioid antagonist, naltrexone alleviates symptoms of AN. This produces an opposite reaction where the perifornical lateral hypothalamus creates an expression of self-stimulation and further promotes the “hunger” response (88).
Pharmacological Perspective
Ciccocioppo R et al. (2004) provides insight into a potential pharmacological drug with anti-anorexic effects (214). Researchers found that neuropeptides, nociceptin/orphanin FQ (N/OFQ) and Ro 64-6198 [synthetic nociceptin (NOP) receptor agonist], exhibit anti-anorexic properties (214). N/OFQ, structurally related to dynorphin A, binds to the NOP receptors in the brain (215,216). When rats were injected three to four micrograms of N/OFQ intracerebroventricularly and two and half milligrams/kilogram of Ro 64-6198 intraperitoneally, they fed at an abnormally high rate (214). Moreover, the effects of N/OFQ along the different sects of the CRF mechanism have been explored. Injecting N/OFQ at the VMH (0.5 Ag/site), the PVNH (0.5 Ag/site), the central nucleus of the amygdala (0.5 Ag/site), the locus coeruleus and the dorsal raphe nucleus (1.0 Ag/rat) demonstrated no change in anorexic behavior (214). However, injecting 0.025-0.25 Ag of N/OFQ in the bed nucleus of the stria terminalis in mice diminished the anorexic behavior (217). Gene knockout experiments performed on mice also demonstrated a high level of reaction to stress in the absence of the N/OFQ gene (218). Moreover, the medial section of bed nucleus of the stria terminalis has been associated with the emotional aspects of stress (214). In conclusion, future drugs should focus on the NOP receptor system with drugs similar to N/OFQ and Ro 64-6195 for the treatment of AN.
Psycho-bio-evolutionary perspective
The mechanism proposed by Yeomans MR et al. (2002) provides a psycho-neurochemical understanding of the opioid system in AN (81). According to the model, AN initially begins with dieting. This leads to a release of opioids and produces a pleasant mood. The second part of the model operates independently and counteractively from the first where the desire to eat inclines, in order to balance the initial self-induced starvation. Finally, the third step involves adapting to starvation by reducing energy output (81). In AN, the first and final steps dominate, so that the individual becomes addicted to dieting and adapts to starvation (88).
Moreover, Davis C et al. (1998) studied the act of self-starvation, aggravated by physical exercise (219). This, itself, is thought to be an addiction to the endogenous opioid system (219). On the Eysenck Personality Questionnaire's addiction scale, anorexics seemed to score high, being similar to the scores of drug addicts and alcoholics. Anorexics also manifested high levels of addictiveness and OCD traits towards weight loss and exercise. The auto-addiction opioid theory hypothesizes that “chronic eating disorders are an addiction to the body's endogenous opioids” (219). Moreover, starvation and excessive physical activity have also shown to increase levels of β-EP, further stimulating dopamine in the mesolimbic reward centers (220,221).
On the other hand, the opioid system involvement in AN has thought to have undergone evolutionary changes. Therefore, this suggests that AN is a result of opioid-mediated mechanisms that have helped animals and humans adapt to short-term food restrictions (81). This mechanism also helps reduce the psychological effects associated with food deprivation.
GHRELIN
During the acute stages of AN, ghrelin levels are distinctly elevated up to two-folds and return to normal levels after weight restoration (95,222-229). Several studies (Figure 1) have demonstrated a negative correlation between BMI and ghrelin levels (95,222,229,230). This reflects a state of negative energy balance. Moreover, fluctuations in the levels of ghrelin are not always influenced by food intake in AN. This suggests some impairment in the regulation of ghrelin (231), perhaps due to chronic adaptation to long-standing food restriction (232).
This diagram shows a gradient relationship between both ghrelin levels and body fat mass in a normal, SOCT and AN patients.
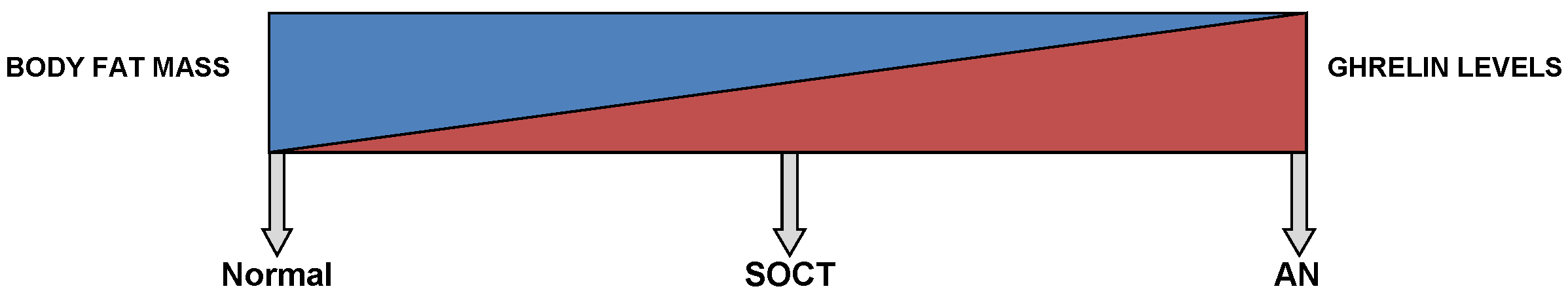
A study by Tolle V et al. (2003) compared ghrelin levels and other nutritional parameters in anorexics and SOCT (229). In AN, patients demonstrated a limited intake of food, BMI <17.5 and reduced body fat, coupled with multiple endocrine changes like hypercortisolism (233,234), hypothyroidism (235), amenorrhea (236), hypoleptinemia (127,237), hyperghrelinemia (91) and alteration of the GH-IGF-1 axis with GH hypersecretion and low IGF-I (238,239). On the contrary, while SOCT and anorexics both displayed parallel BMIs, SOCT underwent normal menstruation and lacked the abnormal feeding behavior. This was further supported by a normal triiodothyronine level, a precise indicator of calorie restriction, in SOCT (229,240,241). In addition, SOCT exhibited results of other endocrine factors (17-β-estradiol, cortisol, GH and IGF-1) similar to the healthy control group. Therefore, these factors are possible indicators of AN (229). Moreover, the normal circadian rhythm in ghrelin secretion has been demonstrated in SOCT (229), as also earlier demonstrated in healthy individuals (94). However, this rhythmicity is absent in AN, with a maximal peak occurring only during the night (229). Moreover, intermediate levels of ghrelin have been observed in SOCT. This signifies that ghrelin partly depends on fat content. This is further supported by the negative correlation (Figure 1) seen between BMI and ghrelin levels (95,223,229).
Moreover, circulating acyl-ghrelin is raised during all phases of an oral glucose tolerance test in AN (222,230). Further studies have described a much higher level of acyl-ghrelin in anorexics than their BMI-matched control group (225-229,242,243). This indicates that persistent hyperghrelinemia possibly impairs ghrelin sensitivity and contributes to the pathogenesis of AN (91). This can be compared to the leptin resistance observed in obesity where subjects are persistently hyperleptinemic (91). Furthermore, persistent hyperghrelinemia possibly impairs the GH/IGF-1 axis, resulting in elevated GH with a paradoxical fall in IGF-1 levels (244-247). Moreover, another study in search of a theory behind “ghrelin resistance” discovered the presence of naturally occurring auto-antibodies to ghrelin (248).
In physiological conditions, these auto-antibodies regulate ghrelin levels in plasma. However, Terashi M et al. (2011) found a significant drop in the levels of acyl-ghrelin immunoglobulin G, immunoglobulin M and immunoglobulin A auto-antibodies in anorexics, persisting to over a month after renutrition (248).
Furthermore, many studies have explored the effects of ghrelin treatment in AN. In a study by Hotta M et al. (2009), six anorexic patients were intravenously infused with three micrograms/kilogram of ghrelin two times daily for fourteen days (249). As a result, energy intake increased by 12-36% with reduced complains of epigastric discomfort and constipation in four patients (249). Also, a significant increase in hunger scores, evaluated by the visual analogue scale, was observed. In another study by Broglio F et al. (2004), a bolus injection of intravenous ghrelin (one microgram/kilogram) brought out a feeling of hunger in six of the nine patients studied (250). In conclusion, ghrelin demonstrated no adverse side effects in the subjects (101,249), but rather it seemed to bring out beneficial changes. An increase in blood glucose levels were observed (251), supporting earlier results suggesting that ghrelin prevented death by maintaining normoglycemia in GOAT -/- mice during periods of starvation (252).
Miljic et al. (2006) studied the effects of prolonged ghrelin infusion, using a five hour protocol, on appetite, sleep and neuroendocrine responses in anorexics (101). As a result, such infusions were unable to bring forth normal GH and appetite responses. However, they suggested that a persistent alteration in the levels of ghrelin and GH response to ghrelin in a partially-recovered anorexic subject, implied persistence of the eating disorder (101). Moreover, increased sleepiness was observed after the fifth hour of infusion (101). In addition, previous studies have demonstrated the role of ghrelin in maintaining slow-wave sleep in humans (253). However, sleep curtailment has shown to limit the secretion of both ghrelin and GH (254-256).
LEPTIN
Leptin exerts its action through binding at two different groups of neurons at the ARCH. The peripheral peptide accesses its receptor (ObRb) through a modified blood brain barrier (257). Binding to the ObRb receptor, the neurons are immediately excited and result in secretion of POMC, a protein that further disintegrates into α-melanocyte stimulating hormone (α-MSH) (258). α-MSH, an anorexigenic neuropeptide, activates the melanocortin-4 (MC4R) and melanocortin-3 (MC3R) receptors and reduces food intake (259,260,261). In addition, secretion of POMC leads to cocaine-and amphetamine-regulated transcript (CART), which further suppresses appetite (262). On the contrary, leptin inhibits the AgRP and NPY neurons, shown to express orexigenic neuropeptides (260). While AgRP has shown to hinder α-MSH/MC4R signaling (261,263), NPY increases food intake and decreases energy loss (264,265). Moreover, the ARCH accounts for only 15-20% of ObRb receptors in the CNS (261,266). Another crucial site for leptin action is at the VMH. Two anorexigenic neuropeptides, steroidogenic factor-1 (SF-1) and brain-derived neurotrophic factor are secreted when leptin binds to the VMH (265,267). SF-1 is a transcription factor essential for the development of the VMH (265,267), while brain-derived neurotrophic factor, a neurotrophin, supports brain growth and controls food consumption (268).
Furthermore, Tolle V et al. (2003) demonstrated significantly low levels of leptin over a twenty-four-hour sampling period in anorexics (229). However, these levels returned to baseline upon renutrition (127,229,237,269-271). As demonstrated in Figure 2, intermediate levels of leptin are found in SOCT, falling in between AN and the healthy control group (229).
This diagram shows a gradient relationship between both leptin levels and body fat mass in a normal, SOCT and AN patients.
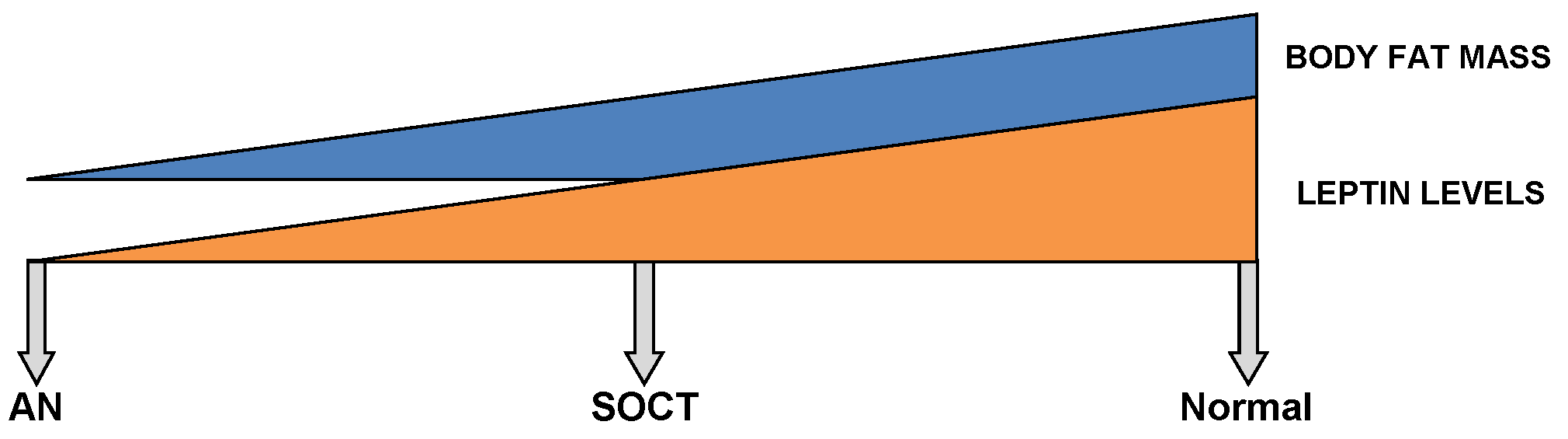
Moreover, body fat mass directly correlates with leptin levels (272,273). Even though anorexics and SOCT follow parallel BMIs, the body composition of the latter group corresponds better with the control. In AN, an excessive diminution of body fat mass is undoubtedly seen. Since SOCT exhibit a greater net body mass than that of anorexics, the intermediate levels of leptin evidently correspond better with SOCT (Figure 2) (229). Moreover, a partial recovery in weight demonstrates an inverse relation between leptin levels and relapse after a one year follow-up (274). In recent weight-recovered anorexics, leptin levels were found to be greater than of their BMI-matched control group. Therefore, this poses difficulties in the further treatment of AN (237,274,275). Moreover, Holtkamp et al. (2003) demonstrated a negative correlation between leptin levels and scores for motor restlessness (276). As a result, pre-clinical and clinical studies have supported hypoleptinemia as the key factor underlying exaggerated physical activity in AN (277).
SEX DIFFERENCES IN THE CRF, OPIOIDS, GHRELIN AND LEPTIN
Understanding the sex differences within the CRF and opioid mechanisms helps stratify their effects in AN. A study by Rivest S et al. (1989) explores the effects of sex differences on energy balance (164). When CRF, representing stress/exercise (278-280), was infused intraventricularly over fourteen days, food intake (protein and fat gain), body weight and energy were reduced in male rats. However, no such changes were seen in females (164). Moreover, the male and female sex hormones, testosterone and estrogen, respectively, are important for mediating CRF and sex differences. The estrogen receptor 1 and estrogen receptor 2 genes, coding for estrogen α and β receptors, are located with CRF and co-regulate its expression (281,282). In addition, Versini A et al. (2010) associated estrogen receptor 1 gene with the RAN subtype (283). Moreover, same-sex and opposite-sex twin studies further support the greater incidence of AN in females (9,15-20). This is probably due to the intrauterine exposure of sex hormones. Also, while estrogen has shown to regulate feeding behavior in females, testosterone has shown minimal effect in males (153).
Administering a selective estrogen α-receptor agonist to ovariectomized rats led to decreased food intake and body weight (284). These agonists also produced varying effects of “social learning of food preference” (285). Furthermore, CRF demonstrates an inhibitory role on gonadotropin-releasing hormone, and subsequently gonadotropin in both sexes. This action is regulated through the opioid-mediated inhibiting action (286-288). Also, CRF reduces estrogen and limits its effect on anorexic behavior in females (289-292), suggesting that low estrogen encourages energy intake (289,293,294). Other studies have suggested that both estrogen and progesterone inhibit feeding under basal (295,296) and inflammatory conditions (297). Estrogen has shown to mediate inhibitory signals for gastric distension and cholecystokinin during digestion (298). Moreover, Miller KK et al. (2005) suggests that testosterone attenuates the symptoms of AN (299), but other studies have demonstrated no effect between low testosterone level and food intake (153,300). In support, Leal et al. (1997) found no relationship between food restriction and diurnal variation of plasma testosterone and androstenedione level in male rats (301). Therefore, the adrenal secretion of corticosterone less likely mediates the diurnal change seen in the male sex hormones. Moreover, many studies have shown sex differences in the sympathetically-driven BAT thermogenesis (301). They found that CRF infusion resulted in high levels of BAT protein in males, but no such effect was seen in females (301).
Sex differences have shown to influence the functioning of the opioid system. This provides insight into why AN is ten to twenty times more prevalent in females (1). Preliminary research on animals demonstrated that µ opioids are more potent in females and κ opioids are more potent in males. However, δ opioids demonstrated similar effects in both sexes (302). Therefore, further research is necessary to understand the sex differences in the effects of µ, κ and δ opioids. Moreover, a pharmacodynamic basis in the sex difference of the opioid mechanism exists. Pharmacodynamic differences include the distribution and density of opioid receptors at different areas of the brain. Research suggests that the male and female hypothalamus exhibit a significant difference in the density of opioid receptors. Accordingly, studies have found higher densities of µ opioids in the male hypothalamus. Gonadal hormones like estrogen have also shown to mediate the levels of opioid and opioid receptor concentration (302).
Studies have shown that females who suffer through chronic illnesses experience early satiety, and present with a high anorexic response related to leptin and tumour necrosis factor- α (303,304). Gayle DA et al. (2006) supported the differential feeding regulation between male and female rats (305). The sex differences in the levels of ghrelin and leptin were studied through administering an orexigenic (calorie restriction) and anorexigenic (inflammatory) stimuli. In both instances, females showed a more positive and stronger response than their male counterparts. In females, the orexigenic stimuli led to increased feeding and high levels of plasma ghrelin, whereas the anorexigenic stimuli only led to a high plasma leptin level. In the inflammatory phenomena, the sex interactions of cytokines, interleukin-1-β and tumour necrosis factor-α with leptin and ghrelin further describe the differential feeding in males and females (305). Accordingly, cytokines have shown to increase leptin (306,307) and decrease ghrelin levels (308). Moreover, basal leptin levels are generally greater in females. Therefore, since high leptin levels are thought to be anorexigenic, this provides insight into why female prevalence is greater in AN.
DISCUSSION
The CRF, opioids, ghrelin and leptin mechanisms operate collectively to demonstrate the underlying physiological and psychological changes in feeding behavior of anorexics (Figure 3). Moreover, these mechanisms have shown to overlap at the HPA axis. These interactions are complex and provide a holistic account for both the physiological and psychological manifestations of AN.
Firstly, the CRF mechanism plays a central role in AN. Literature has suggested that the dysfunction of the CRF mechanism plays a considerable role in the pathogenesis of AN (141). Its actions are broadly distributed within the CNS and PNS, accounting for the various visceral and behavioral manifestations of AN (40). The hyperactivity of the HPA axis, resulting in elevated CRF levels in the CSF (39), has been implicated in the pathogenesis of AN (309). The hyperactivity of the HPA axis results in a negative energy balance, disturbances in sexual function, cardiovascular changes and mood disturbances (147-149). Moreover, CRF1 and CRF2 receptors have shown to mediate the actions of CRF (42-44). In specific, CRF2 receptors have shown to mediate CRF actions of energy intake, independent of the HPA axis (155). The HPA axis-independent pathway functions through the CRF2 receptors, mediated by UCN (156,157). Therefore, CRF regulates energy balance through two independent pathways.
However, it is important to note the effect of each pathway on energy balance. The HPA axis-dependent pathway acts at the central and peripheral level, producing a negative energy balance with activation of the SNS (32,33,73). While, the HPA axis-independent pathway affects energy intake, it lacks peripheral activation (44). The implications of this dual relationship are two-fold. Both pathways have shown to regulate energy balance through CRF and UCN.
However, in support of Baranowska B (1990), the CRF mechanism better accounts for the negative energy balance seen in AN (141). Therapeutically, both pathways need to be fully considered, overlooking either one could result in inadequate treatment of AN.
Irrational and persistent fear is an important component of AN (1). The CRF mechanism plays a central role in forming a fear response (173). There are two HPA-axis related pathways, the independent and dependent pathways that have shown to regulate fear. Mediation of fear and memory formation occurs through an acute stress stimulus (173-175). Moreover, the hippocampus (173), amygdala (175), dorsal raphe (183) and lateral septum (61), work collectively to produce the fear response. The hippocampus helps in the formation of memory, as well as generates a fear response following an acute stress stimulus. At the hippocampus, CRF mediates its actions through both the CRF1 and CRF2 receptors (180-182). In addition to both the psychological (72) and physiological components of the fear response, the amygdala is responsible for arousal, fear and rage reactions through activation of the SNS (310). Therefore, fear conditioning is not fully independent of the HPA axis. The activation of the SNS indicates partial HPA-axis involvement. The effects of amygdala are perhaps mediated through the CRF1 receptors, since the CRF2 receptors have shown to be independent of the HPA axis. The functions of the dorsal raphe (183) and the lateral septum (61) are mediated by the CRF2 receptors. Moreover, the short-term fear response is regulated through the CRF1 receptors while the long-term fear response is regulated through the CRF2 receptors (176). Both receptors have shown to activate the initial signaling pathways (177-179), but only the CRF2 receptors promote associative and stress-related learning (176,180-182). Moreover, the hippocampus is involved in both short-term and long-term effects of fear conditioning, through the action of CRF on both receptors, in the hippocampus. In addition, the hippocampus has shown to consolidate short-term memories into long-term memories (311). CRF also regulates short-term and working memory, seen in fear conditioning, through the CRF1 receptor. Therefore, long-term changes in memory are mediated through UCN, being the predominant agonist to CRF2 receptors. All in all, the hippocampus integrates the actions of both the CRF1 and CRF2 receptors to form durable memories. The interaction of the dorsal raphe and the lateral septum, through the CRF2 receptors, suggests their involvement in the long-term learning process of AN.
Moreover, cortisol represents another CRF-mediated pathway involved in the fear response. This pathway is HPA axis-dependent. Claes SJ (2004) suggests that hypercortisolemia is linked with excessive fear (72). However, it remains unclear if the fear induced by cortisol is qualitatively representative of the fear seen in AN. Based on the scarce evidence in support of cortisol involvement, it is expected that the HPA axis-independent pathway chiefly modulates fear in AN.
The second component of the model is the opioid system (84). We must note the overlap of the opioids and CRF mechanism at the HPA axis, particularly at the PVNH. Opioid peptides regulate CRF through the NA system (312,313). When clonidine stimulates the NA system, a blunting of the β-EP and β-LP secretion is observed (209). This suggests sub-sensitivity at the postsynaptic NA receptor level (209).
The locus coeruleus is involved in the sympathetic stimulation mechanism through the release of NA during stress (314). The locus coeruleus, along with the other bodily systems, help regulate stress (40), and mediate CRF through the action of opioids. Interestingly, starvation inhibits the NA stimulation of CRF, leading to a depressed locus coeruleus (312,313). However, since stress is a component of AN, the locus coeruleus is probably activated. Therefore, the possibilities are two-fold. Firstly, the effect of the locus coeruleus could be biphasic, and secondly, the discharge of NA could be from alternate foci. Since starvation reduces the secretion of ACTH and cortisol through the NA pathway (312,313), and hypercortisolemia has thought to be associated with AN (2), an alternate source of cortisol secretion is expected. Therefore, taking into account the biphasic effect of the locus coeruleus, therapeutic intervention in AN should be cautiously performed.
Hypercortisolemia has shown to suppress the mesolimbic doparminogenic system (172), suggesting the involvement of antagonistic opioids in AN. This may have an effect on hypercortisolemia and mediate the reward-mediated and anorexic behavior. Therefore, high levels of cortisol are probably a result of dysfunctional opioid peptides (141). Moreover, opioid agonists to the µ receptors may help alleviate symptoms of AN related to hypercortisolemia. In addition, N/OFQ and Ro 64-6198 have also demonstrated anti-anorexic effects (214).
Moreover, it is essential that we also consider the reverse and more direct relationship between CRF and opioid peptides. The dysfunctional CRF mechanism seems to directly affect the opioid system. Firstly, this direct link could explain the inhibitory effect of CRF on gonadotropin-releasing hormone through opioid-mediated inhibiting action (286-288). This interaction provides insight into the overlap seen between CRF and opioid peptides. Moreover, Brambilla F et al. (1991) observed a normal secretion of β-EP and β-LP after CRH stimulation in anorexics (209). The reasoning is two-fold. Firstly, this suggests a loss in rhythmicity of opioid secretion due to dysfunctional opioids at the level of hypothalamus/suprahypothalamus. Secondly, this provides insight into the location of the overlap between CRF and opioid peptides.
The regulation of ghrelin adds another dimension to the pathogenesis of AN. It is important to differentiate anorexics from SOCT. Germain N et al. (2007) concluded that SOCT were characterized by high peptide YY concentration, low ghrelin and low-to-normal levels of glucagon-like-peptide-1 and leptin, while anorexics demonstrated a low peptide YY, high ghrelin and low leptin concentration, suggesting an orexigenic adaptive mechanism of appetite regulation in response to low food intake in AN (243). Regardless of an orexigenic profile, anorexics refuse any sort of food intake. This implies that “psychological determinism” plays an important role (243). Moreover, the psycho-behavioral aspects of opioids emphasize the addictiveness of anorexic behavior. Therefore, both addictiveness and the element of fear should be considered in the suppression of the normal physiological response. Current evidence suggests that the physiological component outweighs the psychological component. However, according to the integration model proposed by this paper, the psychological component seems to be an indispensible component of AN.
According to Germain N et al. (2007), SOCT exhibit an equilibrated energy metabolism, while anorexics demonstrate a negative energy balance (243). While anorexics have a constant fear of gaining weight, SOCT put in all efforts towards gaining weight, and often overfeed with the same intent (243,315). Therefore, this suggests that low body weight is not an effective measure of AN. However, measures like body fat content and other nutritional parameters (discussed earlier), may be useful in differentiating the two entities. Moreover, CRF and ghrelin also overlap at the hypothalamus. The high ghrelin levels result in high ACTH levels, and subsequently, hypercortisolemia (233,234). This suggests an additional pathway for fear conditioning. Moreover, the element of fear and its neurophysiology in AN can be understood by three distinct pathways: CRF, cortisol and ghrelin.
Ghrelin dysfunction provides an alternative mechanism in which low estrogen levels result in musculoskeletal disturbances in AN. Ghrelin disturbances are also mediated through the HPA-axis. High levels of GH and low levels of IGF-1 result in a state of catabolism, which helps maintain the leanness of AN (108-110).
Researchers have identified the role of leptin in dysfunctional feeding behavior. Leptin overlaps with CRF at the hypothalamus through NPY (260). Both leptin and opioids are involved in the secretion of the POMC peptide, resulting in the release of α-MSH, CART and β-LP (84,258,262). Leptin regulates energy balance through α-MSH and CART, (258,262) while opioids utilize β-LP (84).
Moreover, evidence shows that ghrelin and leptin function in opposite directions. Ghrelin is orexigenic and adipogenic in action (93,111-113,115,316), while leptin is anorexigenic and supports adipolysis (317,318). These effects are due to the action of NPY/AgRP on ghrelin and leptin receptors in the hypothalamus (319,320). Ghrelin activates the NPY/AgRP neurons (114,316), whereas leptin inhibits them (126,321). Consequently, the negative energy balance seen in AN, reduces leptin levels; while a positive energy balance seen in obesity, increases leptin levels and decreases plasma ghrelin levels (322). Nonetheless, “If ghrelin behaves like an orexigenic factor, the increase in endogenous ghrelin levels in AN could be considered an adaptive mechanism, promoting energy intake and increasing body fat stores in response to a deficit in energy balance” (229). Therefore, endogenous ghrelin levels in AN could be used as a prognostic marker, differentiating a positive outcome from a poor one. In addition to its prognostic value, various studies have demonstrated the therapeutic use of ghrelin in anorexics (101,249-252). Finally, future studies should further evaluate the efficacy of ghrelin in AN.
Differential action of sex hormones gives reasoning to AN being more prevalent in females. In SOCT, physiological gonadal activity is intact, but in anorexics, this activity is absent. The high ghrelin and low leptin levels with abnormal CRH activity has shown to suppress the reproductive system (323-325). Moreover, studies have implicated estrogen in the regulation of energy intake and “social learning of food preference” (285). Also, estrogen has shown to mediate the opioid system and its receptor concentration through the reverse pathway (302). Perhaps, estrogen controls µ receptors and their sex distribution. Females have shown to have a greater concentration of µ receptors in the CNS than their male counterparts (302). Therefore, µ receptors contribute to anorexic behavior as well as to increased female prevalence in AN.
Since µ receptors are involved in reward-mediated behavior (88), it is important to explore the addictiveness and OCD traits of AN. Davis C et al. (1998) provides insight into the understanding of the addictive component through the “auto-addiction opioids theory” (219). Moreover, research has demonstrated associated OCD traits in individuals suffering from AN (219). Therefore, sex hormones like estrogen, which mediate the opioid system are associated with the addictive and OCD traits of AN. As a result, the addictiveness and compulsiveness are probably sex-determined since opioids favor AN in females. Moreover, males may demonstrate the physiological changes of AN similar to females. However, the addictive and OCD attributes of opioid function are perhaps inactive in males due to differential sex distribution of µ receptors. The implications of this are two-fold. Firstly, AN cannot be disassociated from its psychological component. Secondly, the opioid system is a vital component that should be targeted in the treatment of AN. Therefore, males may only manifest the physiological aspects of feeding and never overtly present as AN. This is perhaps due to the masking effect of the regulatory mechanisms present in males. In females, the active psychological component of AN takes the upper hand and prevents the physiological correction from taking place, making the disorder explicit. This notion can be further supported by the resistance observed in the regulation of ghrelin (231). A similar resistance is also seen with leptin levels, which poses difficulties in recovering from AN (91).
Moreover, female dominance in AN can be explained through the leptin mechanism. In general, females demonstrate a higher baseline level of leptin than in males. Since leptin is anorexigenic and supports adipolysis (317,318), this explains the selective sex-dominance in AN.
In sum, it is important to highlight the cause and effect relationship among the different mechanisms of AN. Integrating the various dimensions seen in Figure 3, this would aid clinicians in the management of anorexic patients. Studies have linked HPA-axis activation with starvation (142-145). This association could be an effect of starvation, where starvation activates the HPA-axis and regulates various mechanisms. Brambilla F et al. (1991) further links POMC hypersecretion with starvation (209). Since POMC regulates both leptin and opioids, their involvement in starvation is inevitable. Again, this hypersecretion is an effect of starvation. According to the Yeomans MR et al. (2002) model, initial starvation in AN leads to a release of opioid peptides (81). This induces a pleasant mood, creates an addiction towards dieting and later results in chronic adaptation to starvation (81). Moreover, opiate-addicts and AN patients have key differences in their presentations, this further reinforces that opioids are not causally implicated in AN. Also, there seems to be an overlap with the physical attributes between both groups (203). Most importantly, both groups are physically anorexic; however, the personality attributes of each group differ (203). This supports the atypical functioning of opioids giving sufferers a unique spectrum of clinical manifestations in AN (204).
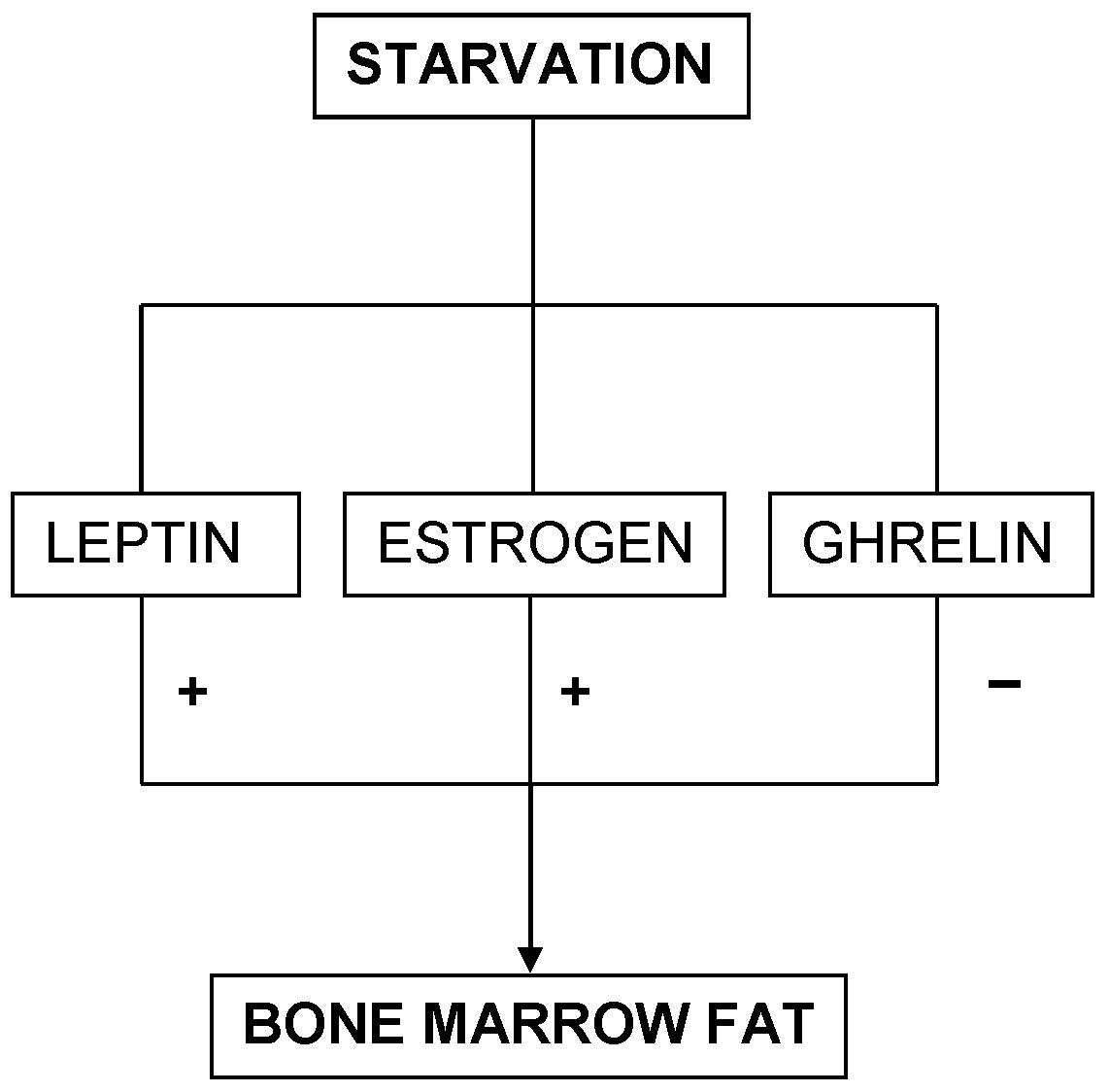
On the other hand, leptin directly correlates with adiposity (127). Devlin MJ (2011) discusses the key role of leptin in regulating bone marrow fat deposition during starvation (22). Studies have found high amounts of marrow fat in ob/ob mice lacking leptin and db/db mice lacking leptin receptors, irrespective of obesity (22). However, leptin treatment in ob/ob mice was shown to reduce bone marrow fat (326-328). Lower leptin levels lead to a persistence of bone marrow fat, because it promotes autophagy by inhibiting the mTOR protein (329,330). The mTOR protein has shown to inhibit autophagy and promote lipogenesis (329,330). Furthermore, bone marrow fat is resistant to lipolysis until depletion of other fat stores occur (22). Syed et al. (2008) have also found high levels of bone marrow fat in post-menopausal women, suggesting that low estrogen levels are associated with high bone marrow fat (331). In conclusion, these experiments highlight the mechanism through which starvation triggers bone marrow fat deposition (22).
On the contrary, mice experiments have demonstrated a deficiency of liver IGF-1 with high levels of GH associated to low levels of bone marrow fat (332). This pattern is similar to the ghrelin level paradigm seen in AN. The implications of these findings are several-fold. Firstly, leptin and estrogen mechanisms of AN function independent of one another. Secondly, since bone marrow fat is protective and increases survival rate during starvation (22), AN mediated through leptin and estrogen seem to be protective, whereas AN mediated through ghrelin has detrimental outcomes (Figure 4). Thirdly, since all mechanisms seem to interact with one another, only certain factors help favor a single mechanism, either the leptin and estrogen mechanism or the ghrelin mechanism, to take precedence. Therefore, future studies should consider exploring the causes of preference in either pathway. Thus, these three mechanisms seem to predict the survival rate for AN. Patients with low levels of leptin and estrogen will perhaps survive longer than those with high ghrelin levels. This also sheds light upon males possibly having a better survival outcome over females in AN.
This diagram portrays the three survival predictors and their relationship with bone marrow fat in starvation.
A Comment on Future Direction
This paper reviewed the recent and historic evidence of various neurological mechanisms involved in the pathogenesis of AN. Most of the evidence gathered came from experiments performed on mice. Experiments on mice help standardize tests and eliminate the element of false information. However, mice can only be used in understanding the biological aspect of AN, since the psychosocial perspective can only be assessed on human subjects. However, obtaining an accurate and truthful history is a challenge encountered with human subjects. Moreover, this paper highlights the intricate relation of the psychological component of AN. Previous experiments have strictly examined the physiological component of AN, like energy balance. Thus, it would be highly inappropriate for us to assume that experiments can induce AN in mice. Also, most studies have isolated single mechanisms and have analyzed their effects. Therefore, it is recommended that future studies explore the interrelation of various mechanisms in AN. Ideally, a cohort study on both prepubertal males and females, showing high levels of CRF, should be performed with observations made at regular intervals to determine the development of AN. This would eliminate the need for extrapolating data from mice onto humans. Finally, future studies should explore the interactions between these mechanisms in post-AN patients.
It is important to understand that starvation does not necessarily imply AN. If two individuals suffering from starvation are compared, the question arises, are both individuals equally likely to develop AN? In underdeveloped countries, where young children suffer from starvation due to a lack of food, it is important to consider the likelihood of these children developing AN later in life. As a matter of fact, forced starvation will rarely develop into AN. This suggests that voluntary and involuntary starvation are distinct entities having unique mechanisms. Traditionally, AN has predominantly affected the western hemisphere. Therefore, it is essential that we inquire whether those that suffer from AN are predisposed to it. Moreover, what causes starvation to evolve into AN needs to be addressed in future studies. It is quite evident that the thin body image portrayed through the media has an important role in AN. The weight loss industries along with the media are very affluent industries, and constantly promote the glorification of being thin. About 47% of girls enrolled between the fifth and the twelfth grades have shown the desire to lose weight as a result of magazine photos (333), while another 69% of girls have agreed that magazines have influenced their image of the ideal body shape (333). Since, young adolescents are constantly being exposed to media, it is vital to explore the psychological and biological components predisposing an individual to develop AN. Moreover, cultural effects seem to intensify the desire to be thin. The western culture has been a forerunner in promoting the thin body image. However, with the western influence percolating, there seems to be a recent increase of AN in the eastern hemisphere. It is also worth mentioning that many religious groups promote the importance of being healthy by staying thin. In conclusion, the adolescents of today are being constantly overwhelmed with the perception of being thin, ultimately, forcing an individual to incorporate this into their self-concept.
Understanding these mechanisms is crucial towards developing newer innovative techniques for the management of AN. Research has predominantly looked at the CRF and opioid mechanisms separately, and have developed drugs that function individually. Therefore, future pharmacological research should integrate knowledge from both systems, and find a common functionality for drugs. This will result in a drug collectively involving both systems and treating a larger array of symptoms. Pharmacological research should further consider the involvement of ghrelin and leptin. Moreover, this paper delineates the various pathways in the manifestation of key symptoms in AN. It is imperative to pharmacologically target all identified pathways to alleviate these symptoms. Since all four mechanisms overlap at the HPA-axis, targeting the HPA-axis, pharmacologically, is beneficial. However, it must be noted that drugs affecting that area would present with a plethora of adverse side effects. Therefore, drugs targeting the HPA-independent pathways should be developed. Although, being specific in action, this would ensure a narrow spectrum of adverse effects. Moreover, since adolescents are greatly affected, various different behavioral techniques should be attempted. Apart from the usual, newer therapies such as provocative therapy, including laughter therapy has been used in the treatment of AN (334). In a recent study on the beneficial effects of laughter, moderate levels of laughter were shown to promote health, while low and high levels demonstrated no effect (335).
Lastly, to better understand the sex differences in AN, future studies should explore AN in those males showing excessive female characteristics. This would help understand the role of sex hormones in AN. Moreover, survival in both sexes should be explored by inducing the various pathways and observing the differences in survival time.
Finally, since many decades AN has been a feeding epidemic in both adolescents and adult females worldwide. However, it is slowly emerging into the developing countries. AN continues to require more investigations and academic inquires in order to achieve a more comprehensive understanding. Therefore, it is imperative that future studies investigate additional neural mechanisms that would account for more of the yet unknown in the field of AN.
Acknowledgements
Thanks to the following individuals for their unconditional support to our paper:
- Mr. Ankush Kadam (Chairman, Mahatma Gandhi Mission's Medical College, India)
- Dr. Adrian RM Upton (Professor of Department of Medicine, Division of Neurology, McMaster University, Canada)
- Dr. Vallabh B. Yadav (Professor and Head of Department, Community Medicine, Mahatma Gandhi Mission's Medical College, India)
- Dr. Ashfaque Ansari (Assistant Professor, Ear Nose Throat, Mahatma Gandhi Mission's Medical College, India).
Thanks to the following individuals for reviewing the manuscript:
- Dr. Adrian RM Upton (Professor of Department of Medicine, Division of Neurology, McMaster University, Canada)
- Dr. Ashfaque Ansari (Assistant Professor, Ear Nose Throat, Mahatma Gandhi Mission's Medical College)
- Mr. Mohammed Merei (Engineer, University of Toronto)
- The two anonymous reviewers who have greatly influenced the outcome of this paper.
Supplementary Material
Abbreviations used in this paper.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Kaplan HI, Sadock BJ. Synopsis of Psychiatry: Behavioral Sciences Clinical Psychiatry, 8th ed. Baltimore, USA: Williams & Wilkins. 1998
2. Kaye WH. Neuropeptide abnormalities in anorexia nervosa. Psychiatry Res. 1996;62:65-74
3. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed, Text Revision. Washington, DC: American Psychiatric Press. 1994
4. Eddy KT, Keel PK, Dorer DJ. et al. Longitudinal comparison of anorexia nervosa subtypes. Int J Eat Disord. 2002;31:191-201
5. Foulon C, Guelfi JD, Kipman A. et al. Switching to the bingeing/purging subtype of anorexia nervosa is frequently associated with suicidal attempts. Eur Psychiatry. 2007;22:513-9
6. Milos G, Spindler A, Hepp U. et al. Suicide attempts and suicidal ideation: links with psychiatric comorbidity in eating disorder subjects. Gen Hosp Psychiatry. 2004;26:129-35
7. Morris J, Twaddle S. Anorexia nervosa. BMJ. 2007;334:894-8
8. Hudson JI, Hiripi E, Pope Jr HG. et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry. 2007;61:348-58
9. Ramoz N, Versini A, Gorwood P. Eating disorders: an overview of treatment responses and the potential impact of vulnerability genes and endophenotypes. Expert Opin Pharmacother. 2007;8:2029-44
10. Harris EC, Barraclough BM. Suicide as an outcome for medical disorders. Medicine (Baltimore). 1994;73:281-96
11. Hoek HW. Incidence, prevalence and mortality of anorexia nervosa and other eating disorders. Curr Opin Psychiatry. 2006;19:389-94
12. Carter JC, Blackmore E, Sutandar-Pinnock K. Relapse in anorexia nervosa: a survival analysis. Psychol Med. 2004;34:671-9
13. Talbot Y. Anorexia nervosa: a lifestyle disorder. Can Fam Physician. 1983;29:553-7
14. Johnson JG, Cohen P, Kasen S. et al. Eating disorders during adolescence and the risk of physical and mental disorders during early adulthood. Arch Gen Psychiatry. 2002;59:545-52
15. Bulik CM, Sullivan PF, Tozzi F. et al. Prevalence, heritability, and prospective risk factors for anorexia nervosa. Arch Gen Psychiatry. 2006;63:305-12
16. Klump KL, Miller KB, Keel PK. et al. Genetic and environmental influences on anorexia nervosa syndromes in a population-based twin sample. Psychol Med. 2001;31:737-40
17. Kortegaard LS, Hoerder K, Joergensen J. et al. A preliminary population-based twin study of self-reported eating disorder. Psychol Med. 2001;31:361-5
18. Mazzeo SE, Mitchell KS, Bulik CM. et al. Assessing the heritability of anorexia nervosa symptoms using a marginal maximal likelihood approach. Psychol Med. 2009;39:463-73
19. Wade TD, Bulik CM, Neale M. et al. Anorexia nervosa and major depression: shared genetic and environmental risk factors. Am J Psychiatry. 2000;157:469-71
20. Gorwood P, Kipman A, Foulon C. The human genetics of anorexia nervosa. Eur J Pharmacol. 2003;480:163-70
21. McCue MD. Starvation physiology: reviewing the different strategies animals use to survive a common challenge. Comp Biochem Physiol A Mol Integr Physiol. 2010;156:1-18
22. Devlin MJ. Why does starvation make bones fat? Am J Hum Biol. 2011;23:577-85
23. Anghel SI, Wahli W. Fat poetry: a kingdom for PPARg. Cell Res. 2007;17:486-511
24. Guerre-Millo M, Rouault C, Poulain P. et al. PPAR-a-null mice are protected from high-fat diet-induced insulin resistance. Diabetes. 2001;50:2809-14
25. Kroetz DL, Yook P, Costet P. et al. Peroxisome proliferator-activated receptor alpha controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J Biol Chem. 1998;273:31581-9
26. Inagaki T, Lin VY, Goetz R. et al. Inhibition of growth hormone signalling by the fasting-induced hormone FGF21. Cell Metab. 2008;8:77-83
27. Fazeli PK, Bredella MA, Misra M. et al. Preadipocyte factor-1 is associated with marrow adiposity and bone mineral density in women with anorexia nervosa. J Clin Endocrinol Metab. 2010;95:407-13
28. Wang T, Hung CC, Randall DJ. The comparative physiology of food deprivation: from feast to famine. Annu Rev Physiol. 2006;68:223-51
29. Dulloo AG, Jacquet J. An adipose-specific control of thermogenesis in body weight regulation. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S22-S29
30. Ellison PT. Energetics and reproductive effort. Am J Hum Biol. 2003;15:342-51
31. Prentice AM. Starvation in humans: evolutionary background and contemporary implications. Mech Ageing Dev. 2005;126:976-81
32. Vale W, Rivier C, Brown MR. et al. Chemical and biological characterization of corticotropin releasing factor. Recent Prog Horm Res. 1983;39:245-70
33. Rivier CL, Plotsky PM. Mediation by corticotropin releasing factor (CRF) of adenohypophysial hormone secretion. Annu Rev Physiol. 1986;48:475-94
34. Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;15:6340-50
35. Fisher LA. Central actions of corticotropin-releasing factor on autonomic nervous activity and cardiovascular functioning. In: (ed.) Chadwick DJ, Marsh J, Ackrill K. CIBA Foundation symposium on corticotropin-releasing factor. Chichester: Wiley. 1993:243-57
36. Koob GF, Heinrichs SC, Pich EM. et al. The role of corticotropin-releasing factor in behavioural responses to stress. In: (ed.) Chadwick DJ, Marsh J, Ackrill K. CIBA Foundation symposium on corticotropin-releasing factor. Chichester: Wiley. 1993:277-95
37. Spiess J, Rivier J, Rivier C. et al. Primary structure of corticotropin-releasing factor from ovine hypothalamus. Proc Natl Acad Sci U S A. 1981;78:6517-21
38. Vale W, Spiess J, Rivier C. et al. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394-7
39. Krahn DD, Gosnell BA, Majchrzak MJ. The anorectic effects of CRH and restraint stress decrease with repeated exposures. Biol Psychiatry. 1990;27:1094-102
40. Aguilera G, Millan MA, Hauger RL. et al. Corticotropin-releasing factor receptors: distribution and regulation in brain, pituitary, and peripheral tissues. Ann N Y Acad Sci. 1987;512:48-66
41. Koob GF. Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry. 1999;46:1167-80
42. Dieterich KD, Lehnert H, De Souza EB. Corticotropin-releasing factor receptors: An overview. Exp Clin Endocrinol Diabetes. 1997;105:65-82
43. Spiess J, Dautzenberg FM, Sydow S. et al. Molecular properties of the CRF receptor. Trends Endocrinol Metab. 1998;9:140-5
44. Cullen MJ, Ling N, Fosterr AC. et al. Urocortin, corticotropin releasing factor-2 receptors and energy balance. Endocrinology. 2001;142:992-9
45. Contarino A, Dellu F, Koob GF. et al. Reduced anxiety-like and cognitive performance in mice lacking the corticotropin-releasing factor receptor 1. Brain Res. 1999;835:1-9
46. Habib KE, Weld KP, Rice KC. et al. Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci U S A. 2000;97:6079-84
47. Heinrichs SC, Lapsansky J, Lovenberg TW. et al. Corticotropin-releasing factor CRF1, but not CRF2, receptors mediate anxiogenic-like behaviour. Regul Pept. 1997;71:15-21
48. Liebsch G, Landgraf R, Engelmann M. et al. Differential behavioral effects of chronic infusion of CRH1 and CRH2 receptor antisense oligonucleotides into the rat brain. J Psychiatr Res. 1999;33:153-63
49. Lundkvist J, Chai Z, Teheranian R. et al. A non peptidic corticotropin-releasing factor receptor antagonist attenuates fever and exhibits anxiolytic-like activity. Eur J Pharmacol. 1996;309:195-200
50. Mansbach RS, Brooks EN, Chen YL. Antidepressant-like effects of CP-154,526, a selective CRF1 receptor antagonist. Eur J Pharmacol. 1997;323:21-6
51. Skutella T, Probst JC, Renner U. et al. Cortocotropin-releasing hormone receptor (type I) antisense targeting reduces anxiety. Neuroscience. 1998;85:795-805
52. Weninger SC, Dunn AJ, Muglia LJ. et al. Stress-induced behaviors require the corticotropin-releasing hormone (CRH) receptor, but not CRH. Proc Natl Acad Sci U S A. 1999;96:8283-8
53. Smith GW, Aubry JM, Dellu F. et al. Corticotropin-releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093-102
54. Timpl P, Spanagel R, Sillaber I. et al. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162-6
55. Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci. 1983;6:269-324
56. Antoni FA, Palkovits M, Makara GB. et al. Immunoreactive corticotropin-releasing hormone in the hypothalamoinfundibular tract. Neuroendocrinology. 1983;36:415-23
57. Kishimoto T, Pearse RV2nd, Lin CR. et al. A sauvagine/corticotropin-releasing factor receptor expressed in heart and skeletal muscle. Proc Natl Acad Sci U S A. 1995;92:1108- 12
58. Kostich WA, Chen A, Sperle K. et al. Molecular identification and analysis of a novel human corticotropin-releasing factor (CRF) receptor: The CRF2gamma receptor. Mol Endocrinol. 1998;12:1077-85
59. Lovenberg TW, Liaw CW, Grigoriadis DE. et al. Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc Natl Acad Sci U S A. 1995;92:836-40
60. Perrin M, Donaldson C, Chen R. et al. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc Natl Acad Sci U S A. 1995;92:2969-73
61. Ho SP, Takahashi LK, Livanov V. et al. Attenuation of fear conditioning by antisense inhibition of brain corticotropin releasing factor-2 receptor. Brain Res Mol Brain Res. 2001;89:29-40
62. Menard J, Treit D. Lateral and medial septal lesions reduce anxiety in the plus maze and probe-burying tests. Physiol Behav. 1996;60:845-53
63. Yadin E, Thomas E, Grishkat HL. et al. The role of the lateral septum in anxiolysis. Physiol Behav. 1993;53:1077-83
64. Sawchenko PE. Evidence for a local site of action for glucocorticoids in inhibiting CRF and vasopressin expression in the paraventricular nucleus. Brain Res. 1987;403:213-23
65. Swanson LW, Sawchenko PE, Rivier J. et al. Organization of ovine corticotropin-releasing factor immunoreactivity cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165-86
66. Fischman AJ, Moldow RL. Extrahypothalamic distribution of CRF-like immunoreactivity in the rat brain. Peptides. 1982;3:149-53
67. Merchenthaler I, Vigh S, Petrusz P. et al. Immunocytochemical localization of corticotropin-releasing factor (CRF) in the rat brain. Am J Anat. 1982;165:385-96
68. Olschowka JA, O'Donohue TL, Mueller GP. et al. The distribution of corticotropin-releasing factor-like immunoreactive neurons in rat brain. Peptides. 1982;3:995-1015
69. Phelps EA. Human emotion and memory: interactions of the amygdala and hippocampal complex. Curr Opin Neurobiol. 2004;14:198-202
70. MacLean PD. Some psychiatric implications of physiological studies on frontotemporal portion of limbic system (visceral brain). Electroencephalogr Clin Neurophysiol. 1952;4:407-18
71. Rivier C, Vale W. Influence of corticotropin-releasing factor on reproductive functions in the rat. Endocrinology. 1984;114:914-21
72. Claes SJ. Trends in molecular medicine. Ann Med. 2004;36:50-61
73. Brown MR, Fisher LA, Spiess J. et al. Corticotropin-releasing factor: actions on the sympathetic nervous system and metabolism. Endocrinology. 1982;111:928-31
74. Kalin NH, Shelton SE, Kraemer GW. et al. Corticotropin-releasing factor administered intraventricularly to rhesus monkeys. Peptides. 1983;4:217-20
75. Muglia L, Jacobson L, Dikkes P. et al. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature. 1995;373:427-32
76. Valentino RJ, Foote SL, Aston-Jones G. Corticotropin-releasing factor activates noradrenergic neurons of the locus coeruleus. Brain Res. 1983;270:363-7
77. Hashimoto K, Murakami K, Hattori T. et al. Corticotropin-releasing factor (CRF)-like immunoreactivity in the adrenal medulla. Peptides. 1984;5:707-11
78. Suda T, Tomori N, Tozawa F. et al. Immunoreactive corticotropin and corticotropin-releasing factor in human hypothalamus, adrenal, lung cancer, and pheochromocytoma. J Clin Endocrinol Metab. 1984;58:919-24
79. Petrusz P, Merchenthaler I, Maderdrut JL. et al. Central and peripheral distribution and corticotropin-releasing factor. Fed Proc. 1985;44:229-35
80. Udelsman R, Harwood JP, Millan MA. et al. Functional corticotropin-releasing factor receptors in the primate peripheral sympathetic nervous system. Nature. 1986;319:147-50
81. Yeomans MR, Gray RW. Opioid peptides and the control of human ingestive behaviour. Neurosci Biobehav Rev. 2002;26:713-28
82. Morley JE, Blundell JE. The neurobiological basis of eating disorders: some formulations. Biol Psychiatry. 1988;23:53-78
83. Koneru A, Satyanarayana S, Rizwan S. Endogenous opioids: their physiological role and receptors. Global J Pharmacol. 2009;3:149-53
84. Monteleone P. New frontiers in endocrinology of eating disorders. Curr Top Behav Neurosci. 2011;6:189-208
85. Glass MJ, Billington CJ, Levine AS. Opioids and food intake: distributed functional neural pathways? Neuropeptides. 1999;33:360-8
86. Kelley AE, Bakshi VP, Haber SN. et al. Opioid modulation of taste hedonics within the ventral striatum. Physiol Behav. 2002;76:365-77
87. Will MJ, Franzblau EB, Kelley AE. Nucleus accumbens mu-opioids regulate intake of high-fat diet via activation of a distributed brain network. J Neurosci. 2003;23:2882-8
88. Chabane N, Leboyer M, Mouren-Simeoni MC. Opiate antagonists in children and adolescents. Eur Child Adolesc Psychiatry. 2000;9(Suppl 1):I44-I50
89. Sakata I, Nakamura K, Yamazaki M. et al. Ghrelin-producing cells exist as two types of cells, closed and opened-type cells, in the rat gastrointestinal tract. Peptides. 2002;23:531-6
90. Kojima M, Hosoda H, Date Y. et al. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656-60
91. Müller TD, Perez-Tilve D, Tong J. et al. Ghrelin and its potential in the treatment of eating/wasting disorders and cachexia. J Cachex Sarcopenia Muscle. 2010;1:159-67
92. Williams DL, Cummings DE. Regulation of ghrelin in physiologic and pathophysiologic states. J Nutr. 2005;135:1320-5
93. Tschöp M, Wawarta R, Riepl RL. et al. Post-prandial decrease of circulating human ghrelin levels. J Endocrinol Invest. 2001;24:RC19-21
94. Cummings DE, Purnell JQ, Frayo RS. et al. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714-9
95. Ariyasu H, Takaya K, Tagami T. et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J Clin Endocrinol Metab. 2001;86:4753-8
96. Hosoda H, Kojima M, Mizushima T. et al. Structural divergence of human ghrelin. Identification of multiple ghrelin-derived molecules produced by post-translational processing. J Biol Chem. 2003;278:64-70
97. Yang J, Zhao TJ, Goldstein JL. et al. Inhibition of ghrelin O-acyltransferase (GOAT) by octanoylated pentapeptides. Proc Natl Acad Sci U S A. 2008;105:10750-5
98. Gutierrez JA, Solenberg PJ, Perkins DR. et al. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl Acad Sci U S A. 2008;105:6320-5
99. Hillman JB, Tong J, Tschop M. Ghrelin biology and its role in weight-related disorders. Discov Med. 2011;11:521-8
100. Howard AD, Feighner SD, Cully DF. et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273:974-7
101. Miljic D, Pekic S, Djurovic M. et al. Ghrelin has partial or no effect on appetite, growth hormone, prolactin, and cortisol release in patients with anorexia nervosa. J Clin Endocrinol Metab. 2006;91:1491-5
102. Cummings DE, Frayo RS, Marmonier C. et al. Plasma ghrelin levels and hunger scores in humans initiating meals voluntarily without time- and food-related cues. Am J Physiol Endocrinol Metab. 2004;287:E297-304
103. Kamegai J, Tamura H, Shimizu T. et al. Central effect of ghrelin, an endogenous growth hormone secretagogue, on hypothalamic peptide gene expression. Endocrinology. 2000;141:4797-800
104. Kamegai J, Tamura H, Shimizu T. et al. Chronic central infusion of ghrelin increases hypothalamic neuropeptide Y and Agouti-related protein mRNA levels and body weight in rats. Diabetes. 2001;50:2438-43
105. Morton GJ, Cummings DE, Baskin DG. et al. Central nervous system control of food intake and body weight. Nature. 2006;443:289-95
106. Arvat E, Maccario M, Di Vito L. et al. Endocrine activities of ghrelin, a natural growth hormone secretagogue (GHS), in humans: comparison and interactions with hexarelin, a nonnatural peptidyl GHS, and GH-releasing hormone. J Clin Endocrinol Metab. 2001;86:1169-74
107. Peino R, Baldelli R, Rodriguez-Garcia J. et al. Ghrelin-induced growth hormone secretion in humans. Eur J Endocrinol. 2000;143:R11-4
108. Amato G, Carella C, Fazio S. et al. Body composition, bone metabolism, and heart structure and function in growth hormone (GH)-deficient adults before and after GH replacement therapy at low doses. J Clin Endocrinol Metab. 1993;77:1671-6
109. Fuller J, Mynett JR, Sugden PH. Stimulation of cardiac protein synthesis by insulin-like growth factors. Biochem J. 1992;282:85-90
110. Moller N, Vendelbo MH, Kampmann U. et al. Growth hormone and protein metabolism. Clin Nutr. 2009;28:597-603
111. Wren AM, Small CJ, Ward HL. et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology. 2000;141:4325-8
112. Wren AM, Small CJ, Abbott CR. et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes. 2001;50:2540-7
113. Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908-13
114. Shintani M, Ogawa Y, Ebihara K. et al. Ghrelin, an endogenous growth hormone secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes. 2001;50:227-32
115. Wren AM, Seal LJ, Cohen MA. et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992
116. Neary NM, Small CJ, Wren AM. et al. Ghrelin increases energy intake in cancer patients with impaired appetite: acute, randomized, placebo-controlled trial. J Clin Endocrinol Metab. 2004;89:2832-6
117. Hosoda H, Kojima M, Matsuo H. et al. Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem Biophys Res Commun. 2000;279:909-13
118. Tolle V, Zizzari P, Tomasetto C. et al. In vivo and in vitro effects of ghrelin/motilin-related peptide on growth hormone secretion in the rat. Neuroendocrinology. 2001;73:54-61
119. Zhang F, Basinski MB, Beals JM. et al. Crystal structure of the obese protein leptin-E100. Nature. 1997;387:206-9
120. Brennan AM, Mantzoros CS. Drug Insight: the role of leptin in human physiology and pathophysiology--emerging clinical applications. Nat Clin Pract Endocrinol Metab. 2006;2:318-27
121. Margetic S, Gazzola C, Pegg GG. et al. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. 2002;26:1407-33
122. Matarese G, Moschos S, Mantzoros CS. Leptin in immunology. J Immunol. 2005;174:3137-42
123. Sinha MK, Ohannesian JP, Heiman ML. et al. Nocturnal rise of leptin in lean, obese, and non-insulin dependent diabetes mellitus subjects. J Clin Invest. 1996;97:1344-7
124. Licinio J, Mantzoros C, Negrão AB. et al. Human leptin levels are pulsatile and inversely related to pituitary-adrenal function. Nat Med. 1997;3:575-9
125. Bluher S, Mantzoros CS. Leptin in humans: lessons from translational research. Am J Clin Nutr. 2009;89:991S-997S
126. Schwartz MW, Seeley RJ, Campfield LA. et al. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101-6
127. Grinspoon S, Gulick T, Askari H. et al. Serum leptin levels in women with anorexia nervosa. J Clin Endocrinol Metab. 1996;81:3861-3
128. Dardeno TA, Chou SH, Moon HS. et al. Leptin in human physiology and therapeutics. Front Neuroendocrinol. 2010;31:377-93
129. Boden G, Chen X, Mozzoli M. et al. Effect of fasting on serum leptin in normal human subjects. J Clin Endocrinol Metab. 1996;81:3419-23
130. Chan JL, Heist K, DePaoli AM. et al. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J Clin Invest. 2003;111:1409-21
131. Kiess W, Petzold S, Töpfer M. et al. Adipocytes and adipose tissue. Best Pract Res Clin Endocrinol Metab. 2008;22:135-53
132. Rosenbaum M, Nicolson M, Hirsch J. et al. Effects of gender, body composition, and menopause on plasma concentrations of leptin. J Clin Endocrinol Metab. 1996;81:3424-7
133. Saad MF, Damani S, Gingerich RL. et al. Sexual dimorphism in plasma leptin concentration. J Clin Endocrinol Metab. 1997;82:579-84
134. Montague CT, Prins JB, Sanders L. et al. Depot- and sex-specific differences in human leptin mRNA expression: implications for the control of regional fat distribution. Diabetes. 1997;46:342-7
135. Fei H, Okano HJ, Li C. et al. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A. 1997;94:7001-5
136. Lee GH, Proenca R, Montez JM. et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632-5
137. Bjørbaek C, Elmquist JK, Michl P. et al. Expression of leptin receptor isoforms in rat brain microvessels. Endocrinology. 1998;139:3485-91
138. Hileman SM, Pierroz DD, Masuzaki H. et al. Characterization of short isoforms of the leptin receptor in rat cerebral microvessels and of brain uptake of leptin in mouse models of obesity. Endocrinology. 2002;143:775-83
139. Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093-6
140. Elmquist JK, Bjørbaek C, Ahima RS. et al. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535-47
141. Baranowska B. Are disturbances in opioids and adrenergic systems involved in the hormonal dysfunction of anorexia nervosa? Psychoneuroendocrinology. 1990;15:371-9
142. Kaiyala KJ, Woods SC, Schwartz MW. New model for the regulation of energy balance and adiposity by the central nervous system. Am J Clin Nutr. 1995;62(Suppl 5):1123S-1134S
143. Schwartz MW, Figlewicz DP, Baskin DG. et al. Insulin and the central regulation of the energy balance: update 1994. Endocrine Monogr Rev. 1994;2:109-13
144. Campfield LA, Smith FJ, Burn P. The OB protein (leptin) pathway-- a link between adipose tissue mass and central neural networks. Horm Metab Res. 1996;28:619-32
145. Seeley RJ, Matson CA, Chavez M. et al. Behavioral, endocrine, and hypothalamic responses to involuntary overfeeding. Am J Physiol. 1996;271:R819-23
146. Schlesser MA, Winokur G, Sherman BM. Hypothalamic-pituitary-adrenal axis activity in depressive illness. Its relationship to classification. Arch Gen Psychiatry. 1980;37:737-43
147. Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev. 1990;15:71-100
148. Glowa JR, Gold PW. Corticotropin-releasing hormone produces profound anorexigenic effects in the rhesus monkey. Neuropeptides. 1991;18:55-61
149. Arase K, York DA, Shimizu H. et al. Effects of corticotropin-releasing factor on food intake and brown adipose tissue thermogenesis in rats. Am J Physiol. 1988;255:E255-9
150. Morley JE, Levine AS. Corticotrophin-releasing factor, grooming and ingestive behavior. Life Sci. 1982;31:1459-64
151. Bornstein SR, Webster EL, Torpy DJ. et al. Chronic effects of a nonpeptide corticotropin-releasing hormone type I receptor antagonist on pituitary-adrenal function, body weight, and metabolic regulation. Endocrinology. 1998;139:1546-55
152. Buwalda B, de Boer S, Van Kalkeren AA. et al. Physiological and behavioral effects of chronic intracerebroventricular infusion of corticotropin-releasing factor in the rat. Psychoneuroendocrinology. 1997;22:297-309
153. Rivest S, Deshaies Y, Richard D. Effects of corticotropin-releasing factor on energy balance in rats are sex dependent. Am J Physiol. 1989;257:R1417-22
154. Rohner-Jeanrenaud F, Walker CD, Greco-Perotto R. et al. Central corticotropin-releasing factor administration prevents the excessive body weight gain of genetically obese (fa/fa) rats. Endocrinology. 1989;124:733-9
155. Smagin GN, Howell LA, Ryan DH. et al. The role of CRF2 receptors in corticotropin-releasing factor- and urocortin-induced anorexia. Neuroreport. 1998;9:1601-6
156. Donaldson CJ, Sutton SW, Perrin MH. et al. Cloning and characterization of human urocortin. Endocrinology. 1996;137:2167-70
157. Vaughan J, Donaldson C, Bittencourt J. et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature. 1995;378:287-92
158. Bray GA. Regulation of energy balance: studies on genetic, hypothalamic and dietary obesity. Proc Nutr Soc. 1982;41:95-108
159. Holt S, York DA. The effect of adrenalectomy on GDP binding to brown-adipose-tissue mitochondria of obese rats. Biochem J. 1982;208:819-22
160. Romsos DR. Norepinephrine turnover in obese mice and rats. Int J Obes. 1985;2(Suppl 9):55-62
161. Saito M, Bray GA. Adrenalectomy and food restriction in the genetically obese (ob/ob) mouse. Am J Physiol. 1984;246:R20-5
162. Arase K, York DA, Bray GA. Corticosterone inhibition of the intracerebroventricular effect of 2-deoxy-D-glucose on brown adipose tissue thermogenesis. Physiol Behav. 1987;40:489-95
163. Bray GA. Obesity-a disease of nutrient or energy balance? Nutr Rev. 1987;45:33-43
164. Rivest S, Landry J, Richard D. Effects of exercise training on energy balance of orchidectomized rats. Am J Physiol. 1989;257:R550-5
165. Dunn AJ, Berridge CW. Corticotropin-releasing factor administration elicits a stress-like activation of cerebral catecholaminergic systems. Pharmacol Biochem Behav. 1987;27:685-91
166. Emoto H, Tanaka M, Koga C. et al. Corticotropin-releasing factor activates the noradrenergic neuron system in the rat brain. Pharmacol Biochem Behav. 1993;45:419-22
167. Lavicky J, Dunn AJ. Corticotropin-releasing factor stimulates catecholamine release in hypothalamus and prefrontal cortex in freely moving rats as assessed by microdialysis. J Neurochem. 1993;60:602-12
168. Matsuzaki I, Takamatsu Y, Moroji T. The effects of intracerebroventricularly injected corticotropin-releasing factor (CRF) on the central nervous system: behavioral and biochemical studies. Neuropeptides. 1989;13:147-55
169. Jezova D, Ochedalski T, Glickman M. et al. Central corticotropin-releasing hormone receptors modulate hypothalamic-pituitary-adrenocortical and sympathoadrenal activity during stress. Neuroscience. 1999;94:797-802
170. Korte SM, Bouws GA, Bohus B. Central actions of corticotropin-releasing hormone (CRH) on behavioral, neuroendocrine, and cardiovascular regulation: brain corticoid receptor involvement. Horm Behav. 1993;27:167-83
171. Linthorst AC, Flachskamm C, Hopkins SJ. et al. Long-term intracerebroventricular infusion of corticotropin-releasing hormone alters neuroendocrine, neurochemical, autonomic, behavioural and cytokine responses to a systemic inflammatory challenge. J Neurosci. 1997;17:4448-60
172. Pacak K, Tjurmina O, Palkovits M. et al. Chronic hypercortisolemia inhibits dopamine synthesis and turnover in the nucleus accumbens: an in vivo microdialysis study. Neuroendocrinology. 2002;76:148-57
173. Radulovic J, Rühmann A, Liepold T. et al. Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress: differential roles of CRF receptors 1 and 2. J Neurosci. 1999;19:5016-25
174. Shors TJ, Weiss C, Thompson RF. Stress-induced facilitation of classical conditioning. Science. 1992;24:537-9
175. Roozendaal B, Brunson KL, Holloway BL. et al. Involvement of stress-released corticotropin-releasing hormone in the basolateral amygdala in regulating memory consolidation. Proc Natl Acad Sci U S A. 2002;99:13908-13
176. Sananbenesi F, Fischer A, Schrick C. et al. Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory. J Neurosci. 2003;23:11436-43
177. Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030-8
178. Matynia A, Anagnostaras SG, Silva AJ. Weaving the molecular and cognitive strands of memory. Neuron. 2001;32:557-9
179. Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1-10
180. Pedersen WA, Wan R, Zhang P. et al. Urocortin, but not urocortin II, protects cultured hippocampal neurons from oxidative and excitotoxic cell death via corticotropin-releasing hormone receptor type I. J Neurosci. 2002;22:404-12
181. Radulovic M, Hippel C, Spiess J. Corticotropin-releasing factor (CRF) rapidly suppresses apoptosis by acting upstream of the activation of caspases. J Neurochem. 2003;84:1074-85
182. Rossant CJ, Pinnock RD, Hughes J. et al. Corticotropin-releasing factor type 1 and type 2alpha receptors regulate phosphorylation of calcium/cyclic adenosine 3',5'-monophosphate response element-binding protein and activation of p42/p44 mitogen-activated protein kinase. Endocrinology. 1999;140:1525-36
183. Hammack SE, Schmid MJ, LoPresti ML. et al. Corticotropin releasing hormone type 2 receptors in the dorsal raphe nucleus mediate the behavioral consequences of uncontrollable stress. J Neurosci. 2003;23:1019-25
184. Chen Y, Brunson KL, Müller MB. et al. Immunocytochemical distribution of corticotropin-releasing hormone receptor type-1 (CRF(1))-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J Comp Neurol. 2000;420:305-23
185. Van Pett K, Viau V, Bittencourt JC. et al. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191-212
186. De Souza EB, Insel TR, Perrin MH. et al. Corticotropin-releasing factor receptors are widely distributed within the rat central nervous system: an autoradiographic study. J Neurosci. 1985;5:3189-203
187. An introduction to dopamine. Severance K. http://www.macalester.edu/psychology/whathap/UBNRP/Dopamine/intro.html
188. Heinrichs SC, Menzaghi F, Pich EM. et al. Corticotropin-releasing factor in the paraventricular nucleus modulates feeding induced by neuropeptide Y. Brain Res. 1993;611:18-24
189. Brady LS, Smith MA, Gold PW. et al. Altered expression of hypothalamic neuropeptide mRMAs in food-restricted and food-deprived rats. Neuroendocrinology. 1990;52:441-7
190. Haas DA, George SR. Neuropeptide Y administration acutely increases hypothalamic corticotropin-releasing factor immunoreactivity: lack of effect in other rat brain regions. Life Sci. 1987;41:2725-31
191. Härfstrand A. Brain neuropeptide Y mechanisms. Basic aspects and involvement in cardiovascular and neuroendocrine regulation. Acta Physiol Scand Suppl. 1987;565:1-83
192. Morley JE. Neuropeptide regulation of appetite and weight. Endocr Rev. 1987;8:256-87
193. Krahn DD, Gosnell BA, Levine AS. et al. Behavioural effects of corticotropin-releasing factor: localization and characterization of central effects. Brain Res. 1988;443:63-9
194. Pich EM, Messori B, Zoli M. et al. Feeding and drinking responses to neuropeptide Y injections in the paraventricular hypothalamic nucleus of aged rats. Brain Res. 1992;575:265-71
195. Morley JE, Levine AS, Gosnell BA. et al. Effect of neuropeptide Y on ingestive behaviours in the rat. Am J Physiol. 1987;252:R599-609
196. Stanley BG, Leibowitz SF. Neuropeptide Y injected in the paraventricular hypothalamus: a powerful stimulant of feeding behavior. Proc Natl Acad Sci U S A. 1985;82:3940-3
197. Kalra SP, Dube MG, Kalra PS. Continuous intraventricular infusion of neuropeptide Y evokes episodic food intake in satiated female rats: effects of adrenalectomy and cholecystokinin. Peptides. 1988;9:723-8
198. Stanley BG, Lanthier D, Chin AS. et al. Suppression of neuropeptide Y-elicited eating by adrenalectomy or hypophysectomy: reversal with corticosterone. Brain Res. 1989;501:32-6
199. Beck B, Jhanwar-Uniyal M, Burlet A. et al. Rapid and localized alterations of neuropeptide Y in discrete hypothalamic nuclei with feeding status. Brain Res. 1990;528:245-9
200. Kalra SP, Dube MG, Sahu A. et al. Neuropeptide Y secretion increases in the paraventricular nucleus in association with increased appetite for food. Proc Natl Acad Sci U S A. 1991;88:10931-5
201. Sahu A, Kalra PS, Kalra SP. Food deprivation and ingestion induce reciprocal changes in neuropeptide Y concentrations in the paraventricular nucleus. Peptides. 1988;9:83-6
202. Hubner HF. Endorphins, eating disorders, and other addictive behaviours. New York, USA: WW Norton & Company. 1993
203. Abbate-Daga G, Amianto F, Rogna L. et al. Do anorectic men share personality traits with opiate dependent men? A case-control study. Addict Behav. 2007;32:170-4
204. Marrazzi MA, McQuarters A, Barnes C. et al. Male/female comparison of morphine effect on food intake-relation to anorexia nervosa. Pharmacol Biochem Behav. 1996;53:433-5
205. Kaye WH. Neuropeptide abnormalities. In: (ed.) Halmi KA. Psychobiology and treatment of anorexia nervosa and bulimia nervosa. Washington DC: American Psychiatric Press. 1992:169-91
206. Kaye WH, Berrettini WH, Gwirtsman HE. et al. Reduced cerebrospinal fluid levels of immunoreactive pro-opiomelanocortin related peptides (including beta-endorphin) in anorexia nervosa. Life Sci. 1987;41:2147-55
207. Kaye WH, Pickar D, Naber D. et al. Cerebrospinal fluid opioid activity in anorexia nervosa. Am J Psychiatry. 1982;139:643-5
208. Gerner RH, Sharp B. CSF beta-endorphin-immunoreactivity in normal, schizophrenic, depressed, manic and anorexic subjects. Brain Res. 1982;237:244-7
209. Brambilla F, Ferrari E, Petraglia F. et al. Peripheral opioid secretory pattern in anorexia nervosa. Psychiatry Res. 1991;39:115-27
210. Tepper R, Weizman A, Apter A. et al. Elevated plasma immunoreactive beta-endorphin in anorexia nervosa. Clin Neuropharmacol. 1992;15:387-91
211. Lamberts SW, de Lange SA, Stefanko SZ. Adrenocorticotropin-secreting pituitary adenomas originate from the anterior or the intermediate lobe in Cushing's disease: differences in the regulation of hormone secretion. J Clin Endocrinol Metab. 1982;54:286-91
212. Wilkes MM, Watkins WB, Stewart RD. et al. Localization and quantitation of beta-endorphin in human brain and pituitary. Neuroendocrinology. 1980;30:113-21
213. Glass MJ, Billington CJ, Levine AS. Feeding induced by food deprivation is differentially reduced by opioid receptor antisense oligodeoxynucleotide probes in rats. Brain Res. 2003;987:223-32
214. Ciccocioppo R, Cippitelli A, Economidou D. et al. Nociceptin/orphanin FQ acts as a functional antagonist of corticotropin-releasing factor to inhibit its anorectic effect. Physiol Behav. 2004;82:63-8
215. Meunier JC, Mollereau C, Toll L. et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532-5
216. Reinscheid RK, Nothacker HP, Bourson A. et al. Orphanin FQ: a neuropeptide that activates an opioid-like G protein-coupled receptor. Science. 1995;270:792-4
217. Ciccocioppo R, Fedeli A, Economidou D. et al. The bed nucleus is a neuroanatomical substrate for the anorectic effect of corticotropin-releasing factor and for its reversal by nociceptin/orphanin FQ. J Neurosci. 2003;23:9445-51
218. Köster A, Montkowski A, Schulz S. et al. Targeted disruption of the orphanin FQ/nociceptin gene increases stress susceptibility and impairs stress adaptation in mice. Proc Natl Acad Sci U S A. 1999;96:10444-9
219. Davis C, Claridge G. The eating disorders as addiction: a psychobiological perspective. Addict Behav. 1998;23:463-75
220. Bergh C, Södersten P. Anorexia nervosa, self-starvation and the reward of stress. Nat Med. 1996;2:21-2
221. Casper RC. Behavioral activation and lack of concern, core symptoms of anorexia nervosa? Int J Eat Disord. 1998;24:381-93
222. Harada T, Nakahara T, Yasuhara D. et al. Obestatin, acyl ghrelin, and des-acyl ghrelin responses to an oral glucose tolerance test in the restricting type of anorexia nervosa. Biol Psychiatry. 2008;63:245-7
223. Otto B, Cuntz U, Fruehauf E. et al. Weight gain decreases elevated plasma ghrelin concentrations of patients with anorexia nervosa. Eur J Endocrinol. 2001;145:669-73
224. Nakahara T, Harada T, Yasuhara D. et al. Plasma obestatin concentrations are negatively correlated with body mass index, insulin resistance index, and plasma leptin concentrations in obesity and anorexia nervosa. Biol Psychiatry. 2008;64:252-5
225. Germain N, Galusca B, Grouselle D. et al. Ghrelin/obestatin ratio in two populations with low bodyweight: constitutional thinness and anorexia nervosa. Psychoneuroendocrinology. 2009;34:413-9
226. Prince AC, Brooks SJ, Stahl D. et al. Systematic review and meta-analysis of the baseline concentrations and physiologic responses of gut hormones to food in eating disorders. Am J Clin Nutr. 2009;89:755-65
227. Otto B, Tschöp M, Frühauf E. et al. Postprandial ghrelin release in anorectic patients before and after weight gain. Psychoneuroendocrinology. 2005;30:577-81
228. Haas V, Onur S, Paul T. et al. Leptin and body weight regulation in patients with anorexia nervosa before and during weight recovery. Am J Clin Nutr. 2005;81:889-96
229. Tolle V, Kadem M, Bluet-Pajot MT. et al. Balance in ghrelin and leptin plasma levels in anorexia nervosa patients and constitutionally thin women. J Clin Endocrinol Metab. 2003;88:109-16
230. Nakai Y, Hosoda H, Nin K. et al. Short-term secretory regulation of the active form of ghrelin and total ghrelin during an oral glucose tolerance test in patients with anorexia nervosa. Eur J Endocrinol. 2004;150:913-4
231. Nedvidkova J, Krykorkova I, Bartak V. et al. Loss of meal-induced decrease in plasma ghrelin levels in patients with anorexia nervosa. J Clin Endocrinol Metab. 2003;88:1678-82
232. Yi CX, Heppner K, Tschöp MH. Ghrelin in eating disorders. Mol Cell Endocrinol. 2011;340:29-34
233. Boyar RM, Hellman LD, Roffwarg H. et al. Cortisol secretion and metabolism in anorexia nervosa. N Engl J Med. 1977;296:190-3
234. Licinio J, Wong ML, Gold PW. The hypothalamic-pituitary-adrenal axis in anorexia nervosa. Psychiatry Res. 1996;62:75-83
235. Croxson MS, Ibbertson HK. Low serum triiodothyronine (T3) and hypothyroidism in anorexia nervosa. J Clin Endocrinol Metab. 1977;44:167-74
236. Starkey TA, Lee RA. Menstruation and fertility in anorexia nervosa. Am J Obstet Gynecol. 1969;105:374-9
237. Eckert ED, Pomeroy C, Raymond N. et al. Leptin in anorexia nervosa. J Clin Endocrinol Metab. 1998;83:791-5
238. Counts DR, Gwirtsman H, Carlsson LM. et al. The effect of anorexia nervosa and refeeding on growth hormone-binding protein, the insulin-like growth factors (IGFs), and the IGF-binding proteins. J Clin Endocrinol Metab. 1992;75:762-7
239. Stoving RK, Veldhuis JD, Flyvbjerg A. et al. Jointly amplified basal and pulsatile growth hormone (GH) secretion and increased process irregularity in women with anorexia nervosa: indirect evidence for disruption of feedback regulation within the GH-insulin-like growth factor I axis. J Clin Endocrinol Metab. 1999;84:2056-63
240. Suda AK, Pittman CS, Shimizu T. et al. The production and metabolism of 3,5,3-triiodothyronine and 3,3,5-triiodothyronine in normal and fasting subjects. J Clin Endocrinol Metab. 1978;47:1311-9
241. O'Brian JT, Bybee DE, Burman KD. et al. Thyroid hormone homeostasis in states of relative caloric deprivation. Metabolism. 1980;29:721-7
242. Monteleone P, Serritella C, Martiadis V. et al. Plasma obestatin, ghrelin, and ghrelin/obestatin ratio are increased in underweight patients with anorexia nervosa but not in symptomatic patients with bulimia nervosa. J Clin Endocrinol Metab. 2008;93:4418-21
243. Germain N, Galusca B, Le Roux CW. et al. Constitutional thinness and lean anorexia nervosa display opposite concentrations of peptide YY, glucagon-like peptide 1, ghrelin, and leptin. Am J Clin Nutr. 2007;85:967-71
244. Golden NH, Kreitzer P, Jacobson MS. et al. Disturbances in growth hormone secretion and action in adolescents with anorexia nervosa. J Pediatr. 1994;125:655-60
245. Argente J, Caballo N, Barrios V. et al. Multiple endocrine abnormalities of the growth hormone and insulin-like growth factor axis in patients with anorexia nervosa: effect of short- and long-term weight recuperation. J Clin Endocrinol Metab. 1997;82:2084-92
246. Støving RK, Flyvbjerg A, Frystyk J. et al. Low serum levels of free and total insulin-like growth factor I (IGF-I) in patients with anorexia nervosa are not associated with increased IGF-binding protein-3 proteolysis. J Clin Endocrinol Metab. 1999;84:1346-50
247. Misra M, Miller KK, Bjornson J. et al. Alterations in growth hormone secretory dynamics in adolescent girls with anorexia nervosa and effects on bone metabolism. J Clin Endocrinol Metab. 2003;88:5615-23
248. Terashi M, Asakawa A, Harada T. et al. Ghrelin reactive autoantibodies in restrictive anorexia nervosa. Nutrition. 2011;27:407-13
249. Hotta M, Ohwada R, Akamizu T. et al. Ghrelin increases hunger and food intake in patients with restricting-type anorexia nervosa: a pilot study. Endocr J. 2009;56:1119-28
250. Broglio F, Gianotti L, Destefanis S. et al. The endocrine response to acute ghrelin administration is blunted in patients with anorexia nervosa, a ghrelin hypersecretory state. Clin Endocrinol (Oxf). 2004;60:592-9
251. Miljic D, Djurovic M, Pekic S. et al. Glucose metabolism during ghrelin infusion in patients with anorexia nervosa. J Endocrinol Invest. 2007;30:771-5
252. Zhao TJ, Liang G, Li RL. et al. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci U S A. 2010;107:7467-72
253. Weikel JC, Wichniak A, Ising M. et al. Ghrelin promotes slow-wave sleep in humans. Am J Physiol Endocrinol Metab. 2003;284:E407-15
254. Dzaja A, Dalal MA, Himmerich H. et al. Sleep enhances ghrelin levels in healthy subjects. Am J Physiol Endocrinol Metab. 2004;28:E963-7
255. Spiegel K, Rasali E, Penev P. et al. Brief communication: Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med. 2004;141:846-50
256. Van Cauter E, Latta F, Nedeltcheva A. et al. Reciprocal interactions between the GH axis and sleep. Growth Horm IGF Res. 2004;14(Suppl A):S10-7
257. Arora S, Anubhuti. Role of neuropeptides in appetite regulation and obesity. Neuropeptides. 2006;40:375-401
258. Morrison CD. Leptin signaling in brain: A link between nutrition and cognition? Biochim Biophys Acta. 2009;1792:401-8
259. Ste Marie L, Miura GI, Marsh DJ. et al. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc Natl Acad Sci U S A. 2000;97:12339-44
260. Cowley MA, Smart JL, Rubinstein M. et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480-4
261. Myers MG, Cowley MA, Münzberg H. et al. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537-56
262. Rogge G, Jones D, Hubert GW. et al. CART peptides: regulators of body weight, reward and other functions. Nat Rev Neurosci. 2008;9:747-58
263. Ollmann MM, Wilson BD, Yang YK. et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135-8
264. Stanley BG, Leibowitz SF. Neuropeptide Y: stimulation of feeding and drinking by injection into the paraventricular nucleus. Life Sci. 1984;35:2635-42
265. Gao Q, Horvath TL. Neuronal control of energy homeostasis. FEBS Lett. 2008;582:132-41
266. Leshan RL, Björnholm M, Münzberg H. et al. Leptin receptor signaling and action in the central nervous system. Obesity (Silver Spring). 2006;14(Suppl 5):208S-212S
267. Dhillon H, Zigman JM, Ye C. et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191-203
268. Abizaid A, Gao Q, Horvath TL. Thoughts for food: brain mechanisms and peripheral energy balance. Neuron. 2006;51:691-702
269. Casanueva FF, Dieguez C, Popovic V. et al. Serum immunoreactive leptin concentrations in patients with anorexia nervosa before and after partial weight recovery. Biochem Mol Med. 1997;60:116-20
270. Herpertz S, Albers N, Wagner R. et al. Longitudinal changes of circadian leptin, insulin and cortisol plasma levels and their correlation during refeeding in patients with anorexia nervosa. Eur J Endocrinol. 2000;142:373-9
271. Stoving RK, Vinten J, Handberg A. et al. Diurnal variation of the serum leptin concentration in patients with anorexia nervosa. Clin Endocrinol (Oxf). 1998;48:761-8
272. Ferron F, Considine RV, Peino R. et al. Serum leptin concentrations in patients with anorexia nervosa, bulimia nervosa and non specific eating disorders correlate with the body mass index but are independent of the respective disease. Clin Endocrinol (Oxf). 1997;46:289-93
273. Mathiak K, Gowin W, Hebebrand J. et al. Serum leptin levels, body fat deposition, and weight in females with anorexia or bulimia nervosa. Horm Metab Res. 1999;31:274-7
274. Holtkamp K, Hebebrand J, Mika C. et al. High serum leptin levels subsequent to weight gain predict renewed weight loss in patients with anorexia nervosa. Psychoneuroendocrinology. 2004;29:791-7
275. Mantzoros C, Flier JS, Lesem MD. et al. Cerebrospinal fluid leptin in anorexia nervosa: correlation with nutritional status and potential role in resistance to weight gain. J Clin Endocrinol Metab. 2006;82:1845-51
276. Holtkamp K, Herpertz-Dahlmann B, Mika C. et al. Elevated physical activity and low leptin levels co-occur in patients with anorexia nervosa. J Clin Endocrinol Metab. 2003;88:5169-74
277. Van Elburg AA, Kas MJ, Hillebrand JJ. et al. The impact of hyperactivity and leptin on recovery from anorexia nervosa. J Neural Transm. 2007;114:1233-7
278. Charpenet G, Tache Y, Forest MG. et al. Effects of chronic intermittent immobilization stress on rat testicular androgenic function. Endocrinology. 1981;109:1254-8
279. Guezennec CY, Ferre P, Serrurier B. et al. Effects of prolonged physical exercise and fasting upon plasma testosterone level in rats. Eur J Appl Physiol Occup Physiol. 1982;49:159-68
280. Rivier C, Rivier J, Vale W. Stress-induced inhibition of reproductive functions: role of endogenous corticotropin-releasing factor. Science. 1986;231:607-9
281. Dagnault A, Richard D. Involvement of the medial preoptic area in the anorectic action of estrogens. Am J Physiol. 1997;272:R311-7
282. Bao AM, Hestiantoro A, Van Someren EJ. et al. Colocalization of corticotropin-releasing hormone and oestrogen receptor-alpha in the paraventricular nucleus of the hypothalamus in mood disorders. Brain. 2005;128:1301-13
283. Versini A, Ramoz N, Le Strat Y. et al. Estrogen receptor 1 gene (ESR1) is associated with restrictive anorexia nervosa. Neuropsychopharmacology. 2010;35:1818-25
284. Santollo J, Wiley MD, Eckel LA. Acute activation of ER alpha decreases food intake, meal size, and body weight in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol. 2007;293:R2194-201
285. Clipperton AE, Spinato JM, Chernets C. et al. Differential effects of estrogen receptor alpha and beta specific agonists on social learning of food preferences in female mice. Neuropsychopharmacology. 2008;33:2362-75
286. Gindoff PR, Ferin M. Endogenous opioids peptides modulate the effect of corticotropin-releasing factor on gonadotropin release in the primate. Endocrinology. 1987;121:837-42
287. Petraglia F, Sutton S, Vale W. et al. Corticotropin-releasing factor decreases plasma luteinizing hormone levels in female rats by inhibiting gonadotropin-releasing hormone release into hypophysial-portal circulation. Endocrinology. 1987;120:1083-8
288. Petraglia F, Vale W, Rivier C. Opioids act centrally to modulate stress-induced decrease in luteinizing hormone in the rat. Endocrinology. 1986;119:2445-50
289. Hervey E, Hervey GR. The influence of sex hormones on energy balance. In: (ed.) Cioffi LA, James WPT, Van Itallie TB. The body weight regulatory system: normal and disturbed mechanisms. New York: Raven. 1981:345-52
290. Himms-Hagen J. Thermogenesis in brown adipose tissue as an energy buffer. Implications for obesity. N Engl J Med. 1984;311:1549-58
291. LeFeuvre RA, Rothwell NJ, Stock MJ. Activation of brown fat thermogenesis in response to central injection of corticotropin-releasing hormone in the rat. Neuropharmacology. 1987;26:1217-21
292. Lofti M, MacDonald IA, Stock MJ. Energy losses associated with oven-drying and the preparation of rat carcasses for analysis. Br J Nutr. 1976;36:305-9
293. Richard D. Effects of ovarian hormones in energy balance and brown adipose tissue thermogenesis. Am J Physiol. 1986;250:R245-9
294. Wade GN, Gray JM, Bartness TJ. Gonadal influences on adiposity. Int J Obes. 1985;1(Suppl 9):83-92
295. Eckel LA. Estradiol: a rhythmic, inhibitory, indirect control of meal size. Physiol Behav. 2004;82:35-41
296. Varma M, Chai JK, Meguid MM. et al. Effect of estradiol and progesterone on daily rhythm in food intake and feeding patterns in Fischer rats. Physiol Behav. 1999;68:99-107
297. Geary N, Asarian L, Sheahan J. et al. Estradiol-mediated increases in the anorexia induced by intraperitoneal injection of bacterial lipopolysaccharide in female rats. Physiol Behav. 2004;82:251-61
298. Geary N. Estradiol, CCK and satiation. Peptides. 2001;22:1251-63
299. Miller KK, Grieco KA, Klibanski A. Testosterone administration in women with anorexia nervosa. J Clin Endocrinol Metab. 2005;90:1428-33
300. Rothwell NJ, Stock MJ. Energy balance and brown fat in adrenalectomized male, female, and castrated male rats. Metabolism. 1986;35:657-60
301. Leal AM, Moreira AC. Daily variation of plasma testosterone, androstenedione, and corticosterone in rats under food restriction. Horm Behav. 1997;31:97-100
302. Craft RM. Sex differences in opioid analgesia: “from mouse to man”. Clin J Pain. 2003;19:175-86
303. Intebi AD, Garau L, Brusco I. et al. Alzheimer's disease patients display gender dimorphism in circulating anorectic adipokines. Neuroimmunomodulation. 2003;10:351-8
304. Walsh D, Donnelly S, Rybicki L. The symptoms of advanced cancer: relationship to age, gender, and performance status in 1,000 patients. Supportive Care Cancer. 2000;8:175-9
305. Gayle DA, Desai M, Casillas E. et al. Gender-specific orexigenic and anorexigenic mechanisms in rats. Life Sci. 2006;79:1531-6
306. Grunfeld C, Zhao C, Fuller J. et al. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J Clin Invest. 1996;97:2152-7
307. Sarraf P, Frederich RC, Turner EM. et al. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med. 1997;185:171-5
308. Abiko Y, Suzuki H, Masaoka T. et al. Enhanced plasma ghrelin levels in Helicobacter pylori-colonized, interleukin-1-receptor type 1-homozygous knockout (IL-1R1-/-) mice. World J Gastroenterol. 2005;11:4148-53
309. Hotta M, Shibasaki T, Masuda A. et al. The responses of plasma adrenocorticotropin and cortisol to corticotropin-releasing hormone (CRH) and cerebrospinal fluid immunoreactive CRH in anorexia nervosa patients. J Clin Endocrinol Metab. 1986;62:319-24
310. Kaada BR. Stimulation and regional ablation of the amygdaloid complex with reference to functional representations. In: (ed.) Eleftheriou BE. The Neurobiology of the Amygdala. New York: Plenum Press. 1972:205-81
311. Alison Preston. Scientific American. How does short-term memory work in relation to long-term memory? Are short-term daily memories somehow transferred to long-term storage while we sleep? Revised September 2007. http://www.scientificamerican.com/article.cfm?id=experts-short-term-memory-to-long-term
312. Grossman A, Gaillard RC, McCartney P. et al. Opiate modulation of the pituitary-adrenal axis: effects of stress and circadian rhythm. Clin Endocrinol (Oxf). 1982;17:279-86
313. Lane HC, Masur H, Edgar LC. et al. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1983;309:453-8
314. Locus Coeruleus. http://www.caam.rice.edu/~cox/wrap/locuscoeruleus.pdf
315. Bossu C, Galusca B, Normand S. et al. Energy expenditure adjusted for body composition differentiates constitutional thinness from both normal subjects and anorexia nervosa. Am J Physiol Endocrinol Metab. 2007;292:E132-7
316. Nakazato M, Murakami N, Date Y. et al. A role for ghrelin in the central regulation of feeding. Nature. 2001;409:194-8
317. Seeley RJ, van Dijk G, Campfield LA. et al. Intraventricular leptin reduces food intake and body weight of lean rats but not obese Zucker rats. Horm Metab Res. 1996;28:664-8
318. Mistry AM, Swick AG, Romsos DR. Leptin rapidly lowers food intake and elevates metabolic rates in lean and ob/ob mice. J Nutr. 1997;127:2065-72
319. Willesen MG, Kristensen P, Rømer J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology. 1999;70:306-16
320. Mercer JG, Hoggard N, Williams LM. et al. Coexpression of leptin receptor and preproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. J Neuroendocrinol. 1996;8:733-5
321. Stephens TW, Basinski M, Bristow PK. et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. 1995;377:530-2
322. Tschöp M, Weyer C, Tataranni PA. et al. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50:707-9
323. De Souza MJ, Leidy HJ, O'Donnell E. et al. Fasting ghrelin levels in physically active women: relationship with menstrual disturbances and metabolic hormones. J Clin Endocrinol Metab. 2004;89:3536-42
324. Holtkamp K, Mika C, Grzella I. et al. Reproductive function during weight gain in anorexia nervosa. Leptin represents a metabolic gate to gonadotropin secretion. J Neural Transm. 2003;110:427-35
325. Norman RL. Corticotropin-releasing hormone effects on luteinizing hormone and cortisol secretion in intact female rhesus macaques. Biol Reprod. 1994;50:949-55
326. Hamrick MW. Leptin, bone mass, and the thrifty phenotype. J Bone Miner Res. 2004;19:1607-11
327. Hamrick MW, Della-Fera MA, Choi YH. et al. Leptin treatment induces loss of bone marrow adipocytes and increases bone formation in leptin-deficient ob/ob mice. J Bone Miner Res. 2005;20:994-1001
328. Hamrick MW, Della Fera MA, Choi YH. et al. Injections of leptin into rat ventromedial hypothalamus increase adipocyte apoptosis in peripheral fat and in bone marrow. Cell Tissue Res. 2007;327:133-41
329. Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542-52
330. Chakrabarti P, English T, Shi J. et al. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes. 2010;59:775-81
331. Syed FA, Oursler MJ, Hefferanm TE. et al. Effects of estrogen therapy on bone marrow adipocytes in postmenopausal osteoporotic women. Osteoporos Int. 2008;19:1323-30
332. Yakar S, Rosen CJ, Bouxsein ML. et al. Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. FASEB J. 2009;23:709-19
333. Levine M. Prevention of Eating Problems with Elementary Children. US: USA Today (July 1998). 1998
334. Provocative Therapy FAQs. Kaplan, Brian. http://drkaplan.co.uk/provocative-therapy-practice/faqs-provocative-therapy
335. Hasan H, Hasan TF. Laugh yourself into a healthier person: a cross cultural analysis of the effects of varying levels of laughter on health. Int J Med Sci. 2009;6:200-11
Author biography
Tasneem Fatema Hasan graduated from medical school in May 2011. Throughout her tenure as a medical student she has achieved academic excellence with first class grades in the University examinations. She was awarded the Mahatma Gandhi Mission's scholarship for both academic and research excellence and the “Flower Award” for her excellence in research. Her first research endeavour was a study on laughter, its effects on health and importance in clinical medicine, for which she was invited to present her findings at an international neuroscience conference in China in 2011. She graduated from Rick Hansen Secondary School, Mississauga Canada, where she was an honor roll student throughout her academic tenure, being awarded the University of Toronto “Book Award” for academic excellence and excellence in community leadership. She was also awarded the “Aiming for the Top scholarship” by the Ontario government. She was chosen as the team captain of the design team for the multinational FIRST Robotics Competition, where she led her team to the prestigious “Chairman's Award at the Greater Toronto Regional” and “Regional 1st place winners at the Waterloo Regional”. Currently, she plans on becoming a family practitioner, specializing in the field of pediatrics.
Hunaid Hasan graduated from medical school in May 2011. During his tenure as a medical student, he has strived for academic excellence with first class marks and achieving a distinction on his university examinations. He was awarded the Mahatma Gandhi Mission's scholarship for both academic and research excellence and the “Flower Award” for his excellence in research. His research endeavors have been a published research paper on laughter, its effects on health and importance in clinical medicine, for which he was invited to present at an international neuroscience conference in China in 2011. His other publication was a research-argumentative paper on Nanotechnology-Nanomedicine and Nanoengineering, published by the University of Toronto Press. Currently, he is working on a research paper on Pure Word Deafness at the Hamilton General Hospital, McMaster University, Canada. Prior, he was pursuing a psychology specialist at the University of Toronto, being awarded an academic scholarship from the University of Toronto. He was chosen for the Research Opportunity Program, to pursue research in the field of happiness and emotions. He was selected by GlaxoSmithKline Inc. for their summer student program in their Regulatory Affairs department. He graduated from Rick Hansen Secondary School, Mississauga Canada with honors. Being 1 of 20 students selected from Canada for the Clarica Scholars Program at the University of Waterloo, he was involved in designing a software tool to help students across the school board write better essays. He was awarded the “STORM Award”, for excellence in academics and profound involvement in the community, the “Rick Hansen Award” for excellence in community leadership, and University of Toronto “Book Award” for excellence in leadership and academic abilities. Currently, he plans on pursuing a residency in clinical medicine and hopes to further pursue his research work in neurology.
![]() Corresponding author: hunaidhasancom or zainabhasan52com. 1345 Daniel Creek Road, Mississauga, Ontario, L5V1V3, Canada.
Corresponding author: hunaidhasancom or zainabhasan52com. 1345 Daniel Creek Road, Mississauga, Ontario, L5V1V3, Canada.