Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2011; 8(7):564-572. doi:10.7150/ijms.8.564 This issue Cite
Research Paper
Atorvastatin Inhibits Myocardial Cell Apoptosis in a Rat Model with Post-myocardial Infarction Heart Failure by Downregulating ER Stress Response
Xian Jing Song1,2, Chun Yan Yang1, Bin Liu2, Qun Wei1, Melvin T. Korkor1, Jie Yu Liu3, Ping Yang1 ![]()
1. Department of Internal Medicine and Cardiology, China-Japan Union Hospital, Norman Bethune College of Medicine, Jilin University, China;
2. Department of Internal Medicine and Cardiology, Second Hospital, Norman Bethune College of Medicine, Jilin University, China;
3. Department of Internal Medicine and Endocrinology, Second Hospital, Norman Bethune College of Medicine, Jilin University, China
Received 2011-5-30; Accepted 2011-8-2; Published 2011-9-22
Abstract
Objective: To determine the effect of atorvastatin on rat heart failure after myocardial infarction and to investigate the underlying mechanism of atorvastatin-mediated myocardium protection.
Methods: Thirty-eight rats were randomly divided into three groups: a heart failure model group (model group), a heart failure plus atorvastatin treatment group (atorvastatin group) and a sham-surgery group (control group). The rat heart failure model was established by ligation of the left anterior descending of coronary arteries (LADs). Changes in hemodynamics parameters were recorded after the final drug administration. Plasma brain natriuretic peptide (BNP) concentration was determined by enzyme-linked immunosorbent assay (ELISA). Histological diagnosis was achieved by hematoxylin and eosin (H&E) and Masson's trichrome staining. The expressions of 78kDa glucose-regulated protein 78 (GRP78), caspase-12 and C/EBP homologous protein (CHOP, also known as GADD153) in myocardial cells and cultured cardiac myocytes were examined by Western blot. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was used to evaluate myocardial cell apoptosis, and flow cytometry was performed to examine the cell apoptosis of cultured cardiac myocytes.
Results: In the atorvastatin group, myocardial cells were lined up more orderly and myocardial fibrosis level was decreased compared to the model group. The expressions of GRP78, caspase-12 and CHOP in myocardial cells were decreased in atorvastatin group. Moreover, in the atorvastatin-treated group the cell apoptosis rate was reduced and the endoplasmic reticulum (ER) stress was activated in response to heart failure and angiotensin II(Ang II) stimulation.
Conclusions: By down-regulating GRP78, caspase-12 and CHOP expressions in myocardial cells in rat heart failure after myocardial infarction, atorvastatin treatment decreased the apoptosis of myocardial cells, suggesting the possible mechanism by which atorvastatin functions in protecting against heart failure.
Keywords: atorvastatin, heart failure, myocardial infarction, ER stress response, apoptosis.
Introduction
Heart failure (HF) after myocardial infarction (MI) is a common clinical syndrome with high morbidity and mortality [1]. It has been suggested that HF is closely correlated with cardiac remodeling, as the dysfunction of cardiac myocyte activates numerous signaling pathways which ultimately lead to remodeling [1]. Although a variety of known factors are involved in the development and progression of HF, the mechanism of HF after MI is still poorly understood.
Increased cardiomyocyte apoptosis has been observed in HF after MI both in animal models and in humans [2, 3]. Additionally, oxidative stress plays a significant role in tissue necrosis and reperfusion injury during this process [4]. It has been demonstrated that subcellular organelle endoplasmic reticulum (ER) greatly developed in hypertrophic and failing hearts, implying ER dysfunction may contribute to heart failure [5].
In response to various cellular stresses, ER stress response (ERSR) is activated to protect and repair stress-induced cellular disorder by inducing cell apoptosis. Three signaling pathways are involved in cell apoptosis mediated through the ERSR: the C/EBP homologous protein (CHOP), the caspase-12, and the c-Jun NH2-Terminal kinase (JNK) [6-8]. In addition, as a chaperone in the ER, glucose regulated protein 78 (GRP78) plays a crucial role in the regulation of the ER dynamic equilibrium, and is a marker for ERSR [9]. Recent studies showed that ERSR may lead to cardiac myocyte apoptosis during the progression of HF [10, 11]. Under such circumstances, the inhibition of ERSR may suppress the activation of ERSR-induced cell apoptosis signaling pathway, so that heart function will be improved [12].
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins) therapy has been demonstrated to benefit patients with HF,of either ischemic or non-ischemic causes, by improving endothelial function, inhibiting inflammatory cytokines, potentiating nitric oxide (NO) synthesis, restoring impaired autonomic function, reversing pathologic myocardial remodeling, and inhibiting apoptosis of myochardial cells [13]. The application of atorvastatin, a clinical statin drug, in patients with non-ischemic HF improved left ventricular ejection fraction (LVEF) and attenuated adverse left ventricular remodeling [14]. In addition, atorvastatin depressed inflammatory process in patients with HF [15]. Nevertheless, the functional role of atorvastatin in myocardial protection after MI, and whether the impacts of atorvastatin treatment-induced apoptosis inhibition are mediated by interfering with ERSR, has not yet been clearly illustrated.
In the present study, an in vivo rat MI model and angiotensin II (Ang II) - stimulated cardiac myocytes in vitro were established for evaluating the potential effect of atorvastatin on myocardial protection. Our results imply that atorvastatin inhibited apoptosis of myocardial cells in rat HF after MI by reducing the ERSR. This study therefore revealed the possible underlying mechanism by which atorvastatin functions in myocardial protection.
Materials and Methods
Reagents
Atorvastatin was purchased from Pfizer (New York City, NY, USA). The enzyme-linked immunosorbent assay (ELISA) kit for assessing the level of rat brain natriuretic peptide (BNP) was obtained from Adlitteram Diagnostic Laboratories (USA). Antibodies against Caspase-12, GRP78, CHOP and the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) -staining kit were purchased from Roche Applied Science (South San Francisco, California, USA). Ang II was obtained from Sigma (St. Louis, MO, USA).
Establishment of the rat MI model
Wistar rats (female) weighing 200 to 220 g were obtained from the Center for Laboratory Animals, Medical College, Jilin University (Jilin, China). Animals were randomly divided into an untreated HF group (model, n=15), a HF model +atorvastatin treatment group (atorvastatin treatment group, n=16) and a sham opration group (control, n=7).
To establish the HF model after MI, diethylether-anesthetized rats were fixed on the operating table. The thoracic cavity was opened to expose the heart, and the left coronary arteries (LADs) were ligated. In the control group, the sutures were passed under the LADs without ligation. The thoracic cavity was closed immediately after the heart was returned. Animals were housed routinely following surgery.
Four weeks after surgery, rats in the atorvastatin treatment group received 10 mg/kg/d atorvastatin for 4 weeks. Experiments were performed 4 weeks after the start of the drug administration period (i.e., 8 weeks after surgery). All experimental procedures were approved by the Animal Ethics Committee of Jilin University.
Hemodynamic examination
After the final drug administration, rats were anesthetized with 3% sodium pentobarbital (30 mg/kg) by intraperitoneal injection. Catheters were inserted into the left ventricle through the right common carotid artery. Changes in hemodynamics parameters, including the left ventricular end-diastolic pressure (LVEDP), the left ventricular systolic pressure (LVSP), the maximal rate of rise in blood pressure in the ventricular chamber (+dP/dt max) and the maximal rate of decline in blood pressure in the ventricular chamber (-dP/dtmax) were measured and recorded by an 8-channel polygraph system (RM-6000).
Evaluation of the plasma BNP concentration
Rats were euthanized and blood samples were collected from the abdominal aorta. The blood serum was separated and the plasma BNP concentration was determined by ELISA according to manufacturer's instructions (Adlitteram Diagnostic Laboratories, USA).
Histological diagnosis
Myocardium tissues in the left ventricle of sacrificed rats (approximately 2 mm in thickness) were removed. Samples were fixed in 4% pre-cooled paraformaldehyde for 72 h and embedded in paraffin for histological studies. Paraffin-embedded tissues were sectioned into slices ~5 μm thick. Sections were stained with hematoxylin and eosin (H&E). Masson's trichrome stain was performed to assess myocardial fibrosis. Images were visualized under an optical microscope at ×200 magnification.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
The cell apoptosis rate in the myocardium was determined by TUNEL according to the manufacturer's instructions (Roche Applied Science, South San Francisco, California, USA). Six micrographs were randomly selected and the numbers of healthy or apoptotic cardiomyocytes were counted. The percentage of apoptotic cells was considered the percentage of the total numbers of cells that TUNEL-positive.
Primary culture of neonatal rat cardiac myocytes
Newborn female rats (n = 10) were sterilized with 75% alcohol and euthanized. The hearts were removed and dipped into pre-cooled D-Hank's solution. After washing 3X with D-Hank's, the myocardium was cut into one-millimeter cubic pieces. Tissues were then digested with 0.06% trypsin. After centrifugation, cells were resuspended with Dulbecco's modified Eagle's medium and Ham's F-12 nutrient mixture (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) and incubated in a humidified atmosphere with 5% CO2 at 37°C for 90 min. Cardiac myocytes were then purified by differential adhesion. Forty-eight hours later, cells were arrested in the G0 phase through 24 h of serum-free medium incubation. After that, cells were randomly assigned for four groups. In the control group (Cont), cells were maintained in DMEM/F12. In the atorvastatin treatment group (Ator), cells were treated with a final concentration of 10-5 M atorvastatin. For Ang II stimulation, cells were treated with 10-7 M Ang II, with or without 10-5 M atorvastatin (Ang II + Ator and Ang II, respectively).
Flow cytometry analysis
Twenty-four hours after drug treatment, cells were harvested and adjusted to a confluency of 0.5×106 to 1×106 cells/mL. Subsequently, cells were fixed with 70% pre-colded alcohol and stained with propidium iodide (PI). PI-labeled cells were analyzed using flow cytometry.
Western blot
Total protein from myocardium tissues or cells were extracted and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane, blocked and probed sequentially with primary antibodies against GRP78, caspase-12 and CHOP. After incubation in the primary antibody, the membrane was incubated in an appropriate secondary antibody. After washing, the bound antibody complexes were detected using an electrochemiluminescence reagent.
Statistical analysis
Statistical analyses were performed using SPSS 10.0 software. Data were presented as mean ±standard deviation (SD). The difference between two groups was evaluated by un-paired t test. The differences among different groups were determined by Kruskal-Wallis test. A probability (P) - value of < 0.05 was considered statistically significant.
Results
Atorvastatin suppressed heart failure after MI
To investigate the roles of Atorvastatin in protecting HF after MI, we initially examined the rat heart of different groups for morphological changes. Eight weeks after LAD ligation, hearts from rats in the model group presented with abnormal enlarged ventricular cavity (Fig. 1A). Pathological examination showed that the cardiac myocytes exhibited an irregular shape and arrangement with myocardial fibrosis. Moreover, the number of cardiac myocytes was greatly reduced. Notably, 4 weeks of Atorvastatin treatment (atorvastatin treatment group) significantly suppressed the signs of HF in the MI animals (Fig. 1A).
Atorvastatin suppressed HF in the rat with MI (A) Representative images of hearts. Four weeks after drug administration, the heart was removed, and tissue sections were stained with H&E or Masson's trichrome. Hemodynamic index, including ±dP/dtmax (B), LVEDP and LVSP (C), and the levels of plasma BNP (D) were recorded after the final dose. +dP/dtmax, maximal rate of rise in blood pressure in ventricular chamber; -dp/dtmax, maximal rate of declining in blood pressure in ventricular chamber; LVEDP, left ventricular end diastolic pressure; LVSP, left ventricular systolic pressure. Data were presented as mean ± SD. *P < 0.05 compared with untreated MI model group. (×200 magnification)
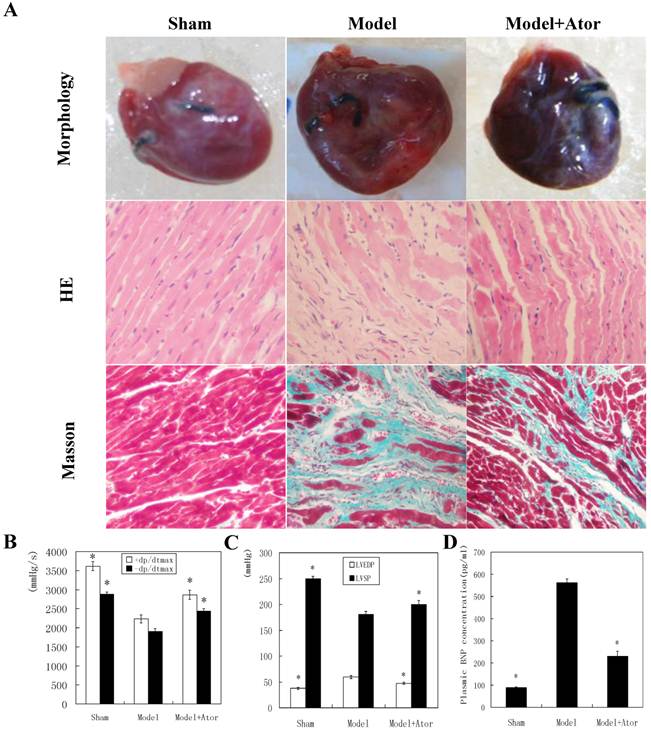
Hemodynamic index was then compared among different groups. As shown in Figure 1, B and C, the untreated model group had increased LVEDP, but decreased LVSP and ± dP/dt max. ELISA analysis showed that the plasma BNP level was promoted in the model group as compared with sham operation group. However, atorvastatin administration significantly reduced the LVEDP and BNP levels, and elevated LVSP and ± dP/dt max levels, compared to the untreated model (P < 0.05). These observations suggested that atorvastatin protected against HF in rats with MI.
Atorvastatin treatment inhibited apoptosis of myocardial cells after MI
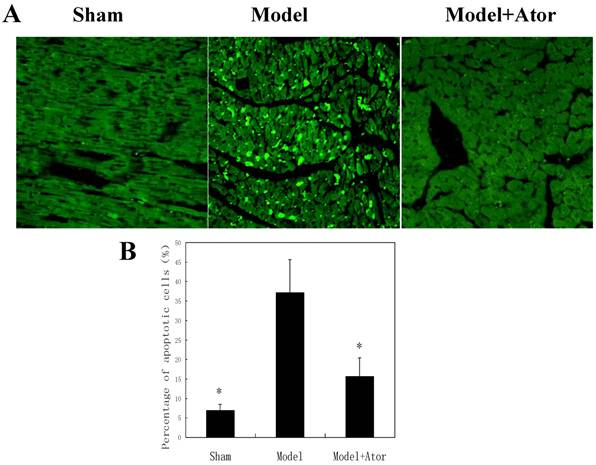
Cell apoptosis is one of the major outcomes of HF after MI, which indicates the molecular mechanism by which the lethality of HF is induced. The observations that atorvastatin protected against HF in rats with MI motivated us to further investigate the impacts of atorvastatin on myocardial cell apoptosis. With the TUNEL assay, we found that increased number of apoptotic myocardial cells were presented in rats with MI induced by LAD ligation, whereas the atorvastatin treatment dramatically decreased cell apoptosis (Fig. 2).
Atorvastatin inhibited cell apoptosis in the rat with MI. TUNEL analysis was carried out four weeks after the end of drug treatments. (A) The TUNEL-positive cells (apoptotic cells) are indicated by arrows. (B) Quantification of apoptotic cell death. *P < 0.05 compared with untreated MI model group.
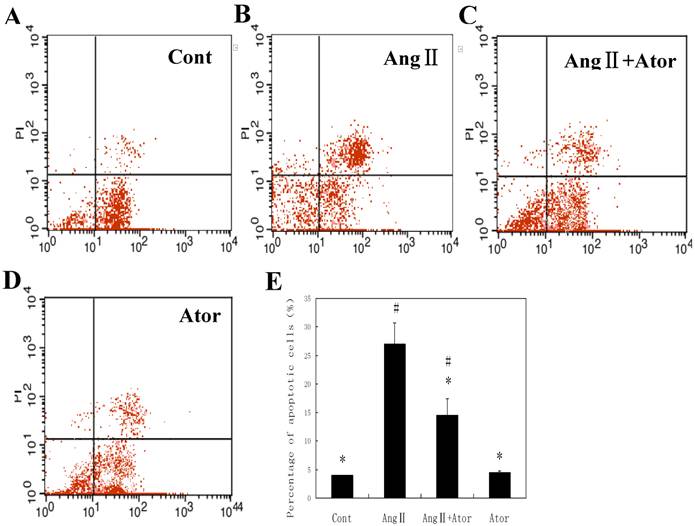
To further confirm this, we then used cultured rat cardiac myocytes to evaluate the effects of atorvastatin. Twenty-four hours after Ang II stimulation, elevated numbers of apoptotic cells labeled by PI staining were detected as revealed, as revealed by flow cytometry (Fig. 3). Atorvastatin greatly suppressed cell apoptosis induced by Ang II. Altogether, these results demonstrated that atorvastatin inhibited cell apoptosis in MI both in vivo and in vitro.
Atorvastatin suppressed cell apoptosis in Ang II-stimulated cardiac myocytes. In the control group (Cont), cells were maintained in DMEM/F12 culture medium. In the atorvastatin treatment group (Ator), cells were incubated with 10-5 M atorvastatin. For Ang II stimulation, cells were treated with 10-7 M Ang II and Ator (Ang II + Ator) or without 10-5 M atorvastatin (Ang II). (A-D) Cell apoptosis was determined by flow cytometry 24 h after drug treatment. Quantified data are presented in (E). *P < 0.05 compared with the Ang II group; #P < 0.05 compared with the Cont group.
Atorvastatin mediated myocardium protection by inhibiting ERSR
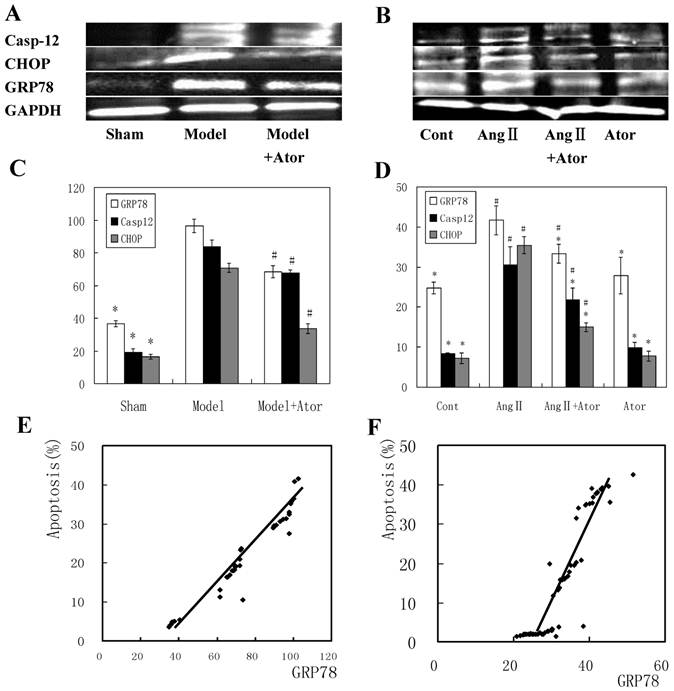
The activation of the ERSR is one of the underlying mechanisms that induce cell apoptosis in HF after MI. To establish the link between ERSR and the inhibition of cell apoptosis induced by atorvastatin, we examined the levels of important proteins involved in ERSR and apoptosis. Among these, GRP78 is a protein of the ER lumen whose expression is induced during ER stress [16]. CHOP has been recognized to play a crucial role in regulating apoptosis induced by ER stress [17], and caspase-12 is required for the apoptotic pathway and contributes to ER stress-triggered caspase activation [18, 19].
As indicated by Fig. 4A-D, elevated levels of GRP78, CHOP and caspase-12 were observed in model group in vivo and in Ang II-stimulated cells in vitro. However, four weeks of atorvastatin administration significantly suppressed all these protein levels, both in vivo and in vitro. Furthermore, the GRP78 level was positively correlated with the cell apoptosis rate both in vivo (r = 0.97694, P < 0.05; Fig.4E) and in vitro r = 0.91578, P < 0.05; Fig. 4F). Collectively, these results demonstrated that the inhibition of cell apoptosis induced by atorvastatin in HF after MI may be linked to the suppression of the ERSR.
Atorvastatin mediated myocardium protection by inhibiting the ERSR. Protein extracted from myocardium tissues four weeks after drug administration (A) or cardiac myocytes 24 h after drug treatment (B) were separated and immunoblotted sequentially with anti-GRP78, -caspase-12 and CHOP antibodies. GAPDH was used as a control reference. Band intensity is quantified in (C) and (D). The correlation between apoptosis and GRP78 is presented in (E) and (F). *P < 0.05 compared with Model or Ang II group; #P < 0.05 compared with sham operated (Sham) or control (Cont) group.
Discussion
A variety of stimuli, such as hypoxia [20, 21], oxidative stress [22], Ang II stimulation [12] and inflammatory factors [23], has been suggested to trigger ER stress during HF. The stressed ER induces the activation of CHOP [23], JNK [24], and caspase-12 [25] and subsequently leads to cardiac myocytes apoptosis. There are evidences that the expressions of GRP78 and CHOP are markedly induced four weeks after transverse aortic constriction (TAC), accompanied by the activation of apoptosis [10]. Heart failure may induce the ERSR, the activated ERSR contributes to cell apoptosis, and finally apoptosis promotes the progression of HF [10].
Consistent with previous studies, here we found elevated expressions of GRP78 and CHOP in vitro and in vivo MI model, suggesting that induction of ER stress occurred during HF after MI. Moreover, the activation of ER stress coincided with the timing of the appearance of apoptosis. Increased TUNEL-positive cells were detected under fluorescent microscope, and elevated numbers of PI-positive cells were found by flow cytometry during HF.
In other studies, caspase-12, which is localized on the ER membrane, was specifically activated by ER stress [18, 19], and in another the cleavage of caspase-12 was not observed in hearts after TAC [10]. However, in the current study, the level of caspase-12 was greatly increased during HF, suggesting the involvement of caspase-12 in ER stress-triggered cell apoptosis. This discrepancy may be due to differences in observed time points or the experimental model.
Atorvastatin, a hydroxymethylglutaryl-coenzyme A reductase inhibitor, has the ability to effectively decrease blood lipids and may provide some advantages over other statins [26]. The administration of atorvastatin was found to improve left ventricular ejection fraction, attenuated adverse left ventricular remodeling in patients with nonischemic HF [14], and depressed the inflammatory process in patients with HF [15]. Nevertheless, the role of atorvastatin in myocardial protection after MI is still poorly known.
In this study, we found that 4 weeks of treatment with atorvastatin dramatically relieved heart failure, as heart morphology was unchanged and hemodynamic indices were maintained in atorvastatin-treated rats. Compared with the model group, atorvastatin administration significantly reduced LVEDP and BNP levels, and elevated LVSP and ±dP/dtmax (P < 0.05). Moreover, atorvastatin decreased the cell apoptosis rate and downregulated the expressions of GRP78, CHOP, and caspase-12 induced by LAD ligation in vivo, and by Ang II stimulation in vitro. This suggests that the myocardial protective effects of atorvastatin were achieved by regulating the ERSR and apoptotic signal pathway.
Although the induction of both the ERSR and apoptosis was observed in the in vivo rat MI model and in the in vitro Ang II-stimulated cardiac myocyte model, the association between ERSR and apoptosis has not been fully demonstrated in this study. There have been reports that the ERSR led to cardiac myocyte apoptosis during the progression of HF [10, 11]. Therefore, it is possible that the ERSR contributes to cell apoptosis in cardiac myocytes. We hypothesize that atorvastatin may inhibit the activation of ER stress by downregulating GRP78 and CHOP expression, which subsequently suppresses the induction of apoptosis. Therefore, our current study presents a novel target for the prevention of MI-induced cardiomyocyte loss. Our future study will be focused on exploring the potential association between the ERSR and apoptosis activation during HF after MI.
Abbreviations
Ang II: angiotensin II; BNP: brain natriuretic peptide; CHOP: C/EBP homologous protein; JNK: c-Jun NH2-Terminal kinase; ER: endoplasmic reticulum; ERSR: endoplasmic reticulum stress response; ELISA: enzyme-linked immunosorbent assay; LADs: left anterior descending of coronary arteries; LVEDP: left ventricular end-diastolic pressure; GRP78: glucose regulated protein 78; FBS: fetal bovine serum; HF: heart failure; H&E: hematoxylin and eosin; Statin: Hydroxymethylglutaryl coenzyme A reductase inhibitor; LVEF: left ventricular ejection fraction; LVEDP: left ventricular end-diastolic pressure; LVSP: left ventricular systolic pressure; +dP/dtmax: maximal rate of rise in blood pressure in ventricular chamber; -dP/dtmax: maximal rate of declining in blood pressure in ventricular chamber; NO: nitric oxide; PI: propidium iodide; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; TAC: transverse aortic constriction; PVDF: polyvinylidene difluoride.
Acknowledgements
This work was supported by grants from the National Science Foundation Item #2009140048 and the Science and Technology Program of Jilin Province, China. Item #200905163.
Conflict of interest
The authors declared no conflict of interest.
References
1. Braunwald E, Bristow MR. Congestive heart failure: fifty years of progress. Circulation. 2000;102(Suppl 4):IV14-23
2. Palojoki E, Saraste A Eriksson A. et al. Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Ph al. 2001;280:H2726-31
3. Saraste A, Pulkki K Kallajoki M. et al. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320-3
4. Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500-8
5. Hamada H, Suzuki M, Yuasa S. et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007-8017
6. Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335-45
7. Breckenridge DG, Germain M, Mathai JP. et al. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608-18
8. Cudna RE, Dickson AJ. Endoplasmic reticulum signaling as a determinant of recombinant protein expression. Biotechnol Bioeng. 2003;81:56-65
9. Chen JC, Wu ML, Huang KC. et al. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc Res. 2008;80:138-50
10. Okada K, Minamino T, Tsukamoto Y. et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705-12
11. Szegezdi E, Duffy A, O'Mahoney ME. et al. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;349:1406-11
12. Wencker D, Chandra M Nquyen K. et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497-504
13. Horwich TB, MacLellan WR Fonarow GC. Statin therapy is associated with improved survival in ischemic and non-ischemic heart failure. J Am Coll Cardiol. 2004;43:642-8
14. Sola S, Mir MQ, Lerakis S. et al. Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure. J Am Coll Cardiol. 2006;47:332-7
15. Tousoulis D, Antoniades C, Bosinakou E. et al. Effects of atorvastatin on reactive hyperemia and inflammatory process in patients with congestive heart failure. Atherosclerosis. 2005;178:359-63
16. Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372-80
17. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381-9
18. Hitomi J, Katayama T Taniquchi M. et al. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci Lett. 2004;357:127-30
19. Nakagawa T, Zhu H, Morishima N. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98-103
20. Paschen W. Endoplasmic reticulum dysfunction in brain pathology: critical role of protein synthesis. Curr Neurovasc Res. 2004;1:173-81
21. Terai K, Hiramoto Y, Masaki M. et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554-75
22. WatanabeY Suzuki O, Haruyama T et al. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem. 2003;89:244-53
23. Wang XZ, Lawson B, Brewer JW. et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol. 1996;16:4273-80
24. Hotamisligil G.S. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54(Suppl 2):S73-8
25. Hetz C, Russelakis-Carneiro M, Maundrell K. et al. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003;22:5435-45
26. Malinowski JM. Atorvastatin: a hydroxymethylglutaryl-coenzyme A reductase inhibitor. Am J Health Syst Pharm. 1998;55:2253-67
Author contact
![]() Corresponding author: Ping Yang, Ph.D, Department of Internal Medicine and Cardiology, China-Japan Union Hospital, Norman Bethune College of Medicine, Jilin University, China. Tel: 0086-431-84995091; Fax: 0086-431-84995091; E-mail: pyangedu.cn.
Corresponding author: Ping Yang, Ph.D, Department of Internal Medicine and Cardiology, China-Japan Union Hospital, Norman Bethune College of Medicine, Jilin University, China. Tel: 0086-431-84995091; Fax: 0086-431-84995091; E-mail: pyangedu.cn.