International Journal of Medical Sciences
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2011; 8(4):287-294. doi:10.7150/ijms.8.287 This issue Cite
Research Paper
Knockdown of NPM1 by RNA Interference Inhibits Cells Proliferation and Induces Apoptosis in Leukemic Cell Line
Feng-Xian Qin1, Hui-Yuan Shao1, Xian-Chun Chen1, Shi Tan1, Hui-Juan Zhang1, Zong-Yu Miao1, Li Wang2, Hui-Chen3, Ling Zhang1 ![]()
1. Key Laboratory of Laboratory Medical Diagnostics, Ministry of Education, Department of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, China.
2. Department of Hematology, the First Affiliated Hospital, Chongqing Medical University, Chongqing 400016, China.
3. Department of Laboratory Medicine, the First Affiliated Hospital, Chongqing Medical University, Chongqing 400016, China.
Received 2011-2-21; Accepted 2011-4-11; Published 2011-4-20
Citation:
Qin FX, Shao HY, Chen XC, Tan S, Zhang HJ, Miao ZY, Wang L, Hui-Chen, Zhang L. Knockdown of NPM1 by RNA Interference Inhibits Cells Proliferation and Induces Apoptosis in Leukemic Cell Line. Int J Med Sci 2011; 8(4):287-294. doi:10.7150/ijms.8.287. https://www.medsci.org/v08p0287.htm
Other stylesAbstract
Nucleophosmin (NPM1) is an abundant and ubiquitously expressed phosphoprotein that is known to influence solid tumors progression. However, little is known about the role of NPM1 in leukemia. Here, we knocked down the NPM1 expression by RNA interference to investigate the role of NPM1 in leukemic cells proliferation and apoptosis. The interference vector pNPM1-shRNA was constructed and transfected into the human leukemic K562 cell line. The expression levels of NPM1 mRNA and protein were detected by quantitative real-time PCR and Western blot, respectively. Cells proliferation potential in vitro was assessed by methyl thiazolyl tetrazolium (MTT) and colony formation assays. Flow cytometry was used to detect the distribution of cell cycle. Cellular apoptosis was reflected by the relative activities of caspase-3 and caspase-8. The results showed that the expression levels of NPM1 mRNA and protein in K562 cells were significantly reduced after pNPM1-shRNA transfection. The cells growth was significantly inhibited in a time-dependent manner and the number of colonies was significantly reduced in the pNPM1-shRNA transfected cells. Meanwhile, the percentage of cells in G1 phase in the K562/pNPM1-shRNA cells was significantly increased. In addition, there were higher relative activities of caspase-3/8 in the pNPM1-shRNA transfected cells. These results indicate that down-regulation of NPM1 expression inhibits leukemic cells proliferation, blocks cell cycle progression and induces cellular apoptosis. It may implicate a potential target for leukemia gene therapy.
Keywords: Nucleophosmin, RNA interference, leukemic cell, proliferation, apoptosis
1. Introduction
Nucleophosmin (NPM1), also known as B23, encoded by the NPM1 gene maps to chromosome band 5q35 in humans [1]. NPM1 is an abundant and ubiquitously expressed phosphoprotein that plays important roles in regulation of multiple cellular functions, such as ribosome biogenesis and transport [2], centrosome duplication [3] and stress responses [4]. In addition, NPM1 is also essential for embryonic development; and it's inactivation in the germ line leads to a series of developmental defects and embryonic lethality at mid-gestation [5, 6]. In recent years, much of interest is paid on NPM1 because this gene implicated in human tumorigenesis. NPM1 is frequently overexpression in solid tumors and has been proposed as a marker for ovarian [7], colon [8], gastric [9], prostate [10] and thyroid tumor [11]. The overexpression of NPM1 also has been detected in leukemia blast and cell lines [12, 13]. In addition, NPM1 is frequently involved in chromosomal translocation in hematological malignancies, forming fusion proteins (NPM1-ALK [14], NPM1-RARa [15] and NPM1-MLF1 [16]). So, the change involved in NPM1 plays an important role in leukemiagenesis.
Accumulating evidences suggest NPM1 protein is one of the key elements in the regulation of nucleolar function. It involves in regulating the susceptibility of leukemia cells to chemotherapeutics or induction of cellular apoptosis. Overexpression of NPM1 was found to decrease the susceptibility of human HL-60 leukemic cells to retinoic acid (RA)-induced apoptosis [17], and to lead to resistance to RA- or TPA-induced differentiation [18], while the down-regulation of NPM1 made the cells more susceptible to BuONa-induced apoptosis [19]. Moreover, NPM1 also plays an important role in cellular mortalization via regulating the activity of telomerase [20]. Recently, some studies indicated that the mutation NPM1 aberrant in the cytoplasm of leukemia blasts would interfere with wild-type NPM1 functions by binding and recruiting them into cytoplasm which may contribute to its leukemiagenesis [21]. In conclusion, the complex behavior of the NPM1 protein demands further studies to better determine the molecular mechanisms and biological consequences of its altered expression in leukemia.
To elucidate the potential role of NPM1 in leukemia, we used RNA interference to knock down the expression of NPM1 in human leukemic K562 cells and detected the effect of NPM1 gene silencing on cells proliferation, cell cycle distribution and cellular apoptosis.
2. Materials and Methods
2.1 Cell line and culture conditions
Human myelogenous leukemic K562 cells (purchased from the Shanghai Institutes for Biological Sciences) were routinely cultured in RPMI 1640 medium (Gibco, MD, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, MD, USA) in a 5% CO2-humidified incubator at 37 °C.
2.2 Construction interference vector
For knock-down of NPM1 genes by RNA interference, the following RNA oligos specific for NPM1 coding region were used: 5'-AATGTCTGTACAGCCAACG-3' [22]. The oligonucleotides for shRNAs were then designed and the structure contained (5' to 3') a BamHI site, the sense sequence (italicized), a loop region, the antisense sequence (italicized), a termination sequence, a ScaI restriction site and a HindⅢ cloning site (shNPM1 sense: 5'-GATCCAATGTCTGTACAGCCAACGTTCAAGACGCGTTGGCTGTACAGACATTTTTTTGTCGAC-3'; antisense: 5'-AGCTTGTCGACAAAAAAATGTCTGTACAGCCAACGCGTCTTGAACGTTGGCTGTACAGACATTG-3'). Oligonucleotides were annealed in 10 mM Tris and 20 mM NaCl (pH 7.6) by heating to 94 °C for 3 min followed by slowly cooling to room temperature. Double-stranded oligonucleotides were then inserted into the plasmid vector pGenesil-1 (Wuhan Genesil Biotechnology Co., Ltd., Wuhan, China) via BamHI and Hind Ⅲ restriction sites. As a negative control, a scrambled sequence of shRNA was constructed by the same strategy. The resulting shRNA expression plasmids were named pNPM1-shRNA and pshRNA-NC, respectively. Both of them can express enhanced green fluorescent protein (GFP). All clones were verified by DNA sequencing.
2.3 Transient transfection
The K562 cells (8.0×105/ml) in logarithmic growth phase were seeded in 6-well plate and transfected with Lipofectamin 2000/pNPM1-shRNA complexes which combined at a 1: 2.5 ratio. The complexes were incubated at room temperature for 20 min, and then added directly to the cells covered in a basic medium. After incubation at 37 °C for 6 h, the complexes were replaced by RPMI 1640 containing 10% FBS. The transfected cells were named as K562/pNPM1-shRNA. The control cells, K562/pshRNA-NC and K562-mock, were established similarly. Each transfection was performed in independent cultures and at different times. When transient transfection was performed for 48 h, the successfully transfected cells with GFP protein were verified by fluorescent microscopy. Cells transfection efficiency is reflected by the percentage of GFP-positive cells.
2.4 Quantitative real-time PCR analysis
Total RNA was extracted from cells in each group, and the quantitative real-time PCR (qRT-PCR) was performed using a SYBR Green reaction (TaKaRa, Tokyo, Japan) kit and a Rotor-Gene 6000 Real-Time PCR machine (Corbett Research, Sydney, Australia). The expression levels of NPM1 were analyzed relative to the levels of the β-actin gene transcript. Sequences of the primers were as follows: NPM1: forward primers, 5'-ACTCCACCCTTTGCTTGGTTT-3' and reverse primers, 5'-TTTGTCTCCCCACCATTTCC-3'; β-actin: forward primers, 5'-AGCGAGCATCCCCCAAAGTT-3' and reverse primers 5'-GGGCACGAAGGCTCATCATT-3'. The reaction mixture for each direct qRT-PCR was performed using 10μl of SYBR Premix Ex Taw, 1 μl (0.4 μM) of each primer and 2 μl DNA, and in a total reaction volume of 20 μl. qRT-PCR reaction condition included predenaturation at 95 °C for 30 sec, followed by 40 cycles of denaturation at 95 °C for 5 sec and annealing at 60 °C for 30 sec. Three independent experiments were performed on the cells in independent cultures at 3 different times.
2.5 Western blot
Cells in each group were washed with ice-cold phosphate-buffered saline (PBS) triplicate and lysed by lysis buffer supplemented with protease inhibitor cocktail. Protein concentrations were determined by BCA method. A total of 50 μg of protein was resolved in 12% sodium dodecyl sulfate-polyacrylamide gel, and then transferred to polyvinylidene difluoride membrane. The member was blocked in 5% low-fat dry milk for 3 h, then was incubated with primary antibody for overnight at 4 °C (anti-NPM1 was used; 1:500 dilution; Santa Cruz Biotechnology, California, USA) and secondary antibody (goat anti-mouse antibody, 1:1 000 dilution, Zhongshan goldenbrioge Biotechnology Co., Ltd., Beijing, China) for 1 h at 37 °C. After triplicate washed with Tris-Buffered Saline Tween-20 (TBST), the autoradiograms obtained were scanned and exported for densitometry analysis. β-actin (anti-β-actin was used, 1:500 dilution; Santa Cruz Biotechnology, California, USA) was used to control the uniform protein content in samples. Three independent experiments were performed on the cells in independent cultures at 3 different times.
2.6 Cells proliferation assay
To measure cells proliferation rate, each group of cells were seeded in 96-well plates at a density of 3×103 cells per well. Cells proliferation was quantified at 0 h, 24 h, 48 h and 72 h with an MTT assay. 20 μl of MTT (5 mg/ml, Sigma, MO, USA) was added to each well and cultured for 4 h at 37 °C and 5% CO2. The liquid medium in the wells was replaced with 150 μl dimethylsulphoxide (DMSO, Sigma, MO, USA), and then the conversion of MTT into DMSO-soluble formazan by living cells was quantified by measuring the absorbance at 490 nm using a spectrophotometer. Cells growth curves were plotted with OD value as ordinate and time as abscissa. Three independent experiments were performed on the cells in independent cultures at 3 different times.
2.7 Colony formation assay
Colony formation assay was performed in semisolid culture medium containing 0.9% methylcellulose (Sigma, MO, USA) and 30% FBS. Each group of cells was seeded in dish at a density of 1×103/ml/dish. After incubating at 37 °C with 5% CO2 for two weeks, routine colony counts were performed under an inverted microscope (XDS-1B, Beijing, China). Aggregates of 50 or more cells were scored as one colony and that of 3~50 cells as one cluster. Three independent experiments were performed on the cells in independent cultures at 3 different times.
2.8 Flow cytometry assay
Each group of cells were washed by PBS for three times, and then fixed with 70% cold ethanol. Cell-cycle analysis was carried out by flow cytometry (Becton Dickinson, CA, USA) after propidium iodide staining. Three independent experiments were performed on the cells in independent cultures at 3 different times.
2.9 Spectrophotometry detection
For determination of caspase-3 activity in the cells, Caspase-3 Assay kit (Clontech, Texas, USA) was used according to the manufacturer's instructions. In brief, cells (4 ×106) in each group were routinely harvested, mixed with 50 μl cellular lysis buffer and then incubated on ice for 30 min. After centrifugation at 4 °C for 1 min, the supernatant (50 μl) was collected and mixed with 50 μl 2× reaction buffer and 5 μl caspase-3 substrate. The complexes was incubation at 37 °C in the dark for 4 h, the optical density values of the samples were read on a spectrophotometer at a wavelength of 400 nm, which represented the intracellular activity of caspase-3. The activity of caspase-8 was measured with the same proposal. Three independent experiments were performed on the cells in independent cultures at 3 different times.
2.10 Statistical analysis
SPSS13.0 software was used for statistical analysis. Results were presented as the mean ± SD for three different determinations. A Student's t-test was used to compare averaged values and values of p<0.05 were considered statistically significant.
3. Results
3.1 Down-regulation of NPM1 in K562 cells with RNAi
To silence NPM1 gene by RNA interference, we transfected pNPM1-shRNA with enhanced GFP into K562 cells. The results revealed that the percentage of GFP-positive cells in each group was 71.5%, 71.2% and 72.7%, respectively (Figure 1A), which indicated that the transfection efficiency was high. qRT-PCR and Western blot analysis showed that both the NPM1 mRNA and protein levels were significantly lower in K562/pNPM1-shRNA cells than those in controls (p<0.05, Figure 1B, C). No significant difference of NPM1 mRNA and protein levels was found among the control groups (p>0.05, Figure 1B, C).
Figure 1
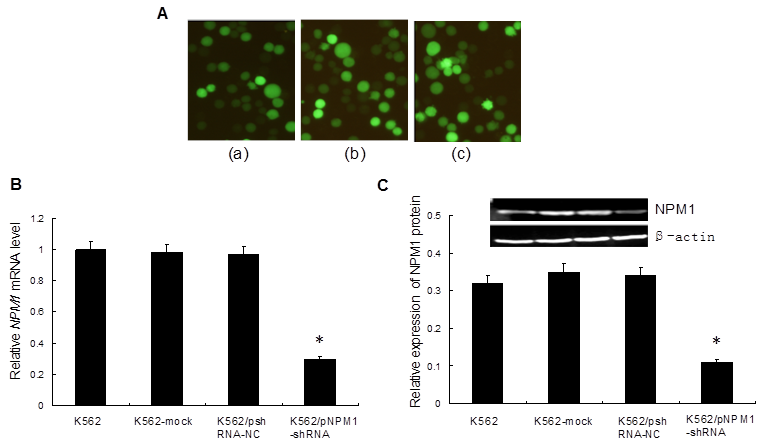
Transfection of the interference vectors and effect of NPM1 gene silencing on NPM1 expression. A, The expression of GFP protein in transfected cells was detected by fluorescence microscopy (×200). (a), K562-mock cells; (b), K562/pshRNA-NC cells; (c), K562/pNPM1-shRNA cells. A representative example of this transfection results is shown. B, The level of NPM1 mRNA expression was detected by qRT-PCR. The expression of NPM1 mRNA in K562/pNPM1-shRNA cells was significantly decreased compared with the control groups. Data are shown as means ± SD of triplicate experiments. *p <0.05. C, The level of NPM1 protein expression was assessed by Western blot. The average signal intensity was standardized to β-actin. The expression of NPM1 protein in K562/pNPM1-shRNA cells was significantly decreased compared with the controls. Data were shown as means ± SD of triplicate experiments. *p <0.05.
3.2 Down-regulation of NPM1 suppressed K562 cells growth
To determine whether down-regulation of NPM1 has an influence on cells growth in vitro, the proliferation of K562 cells was evaluated by an MTT assay. The result showed that, compared with the controls, the proliferation of K562/pNPM1-shRNA cells was significantly inhibited in a time-dependent manner and the highest inhibitory rate was (52.7 ± 2.5%) at 72 h (p < 0.05, Figure 2A). The inhibitory effect of pNPM1-shRNA on K562 cells growth was also confirmed by colony formation assay. As shown in Figure 2B, down-regulation of NPM1 led to 40% reduction of colony numbers in K562/pNPM1-shRNA cells compared with the controls (p<0.05). These data indicated that NPM1 silencing could significantly inhibit K562 cells proliferation.
3.3 Down-regulation of NPM1 arrested K562 cells in the G1 phase
To evaluate the effect of NPM1 gene silencing on the cell cycle progression of K562 cells, the flow cytometry analysis was performed at 48 h after transfection. Results showed that in K562/pNPM1-shRNA group, the percentage of cells in G1 phase was significantly increased, while that in S phase was remarkably decreased (p<0.05, Figure 3 and Table 1). There was no significant difference in cell cycle distribution among the control groups (p>0.05, Figure 3 and Table 1). These data indicated that reduction of NPM1 arrested the cell cycle progression.
Figure 2
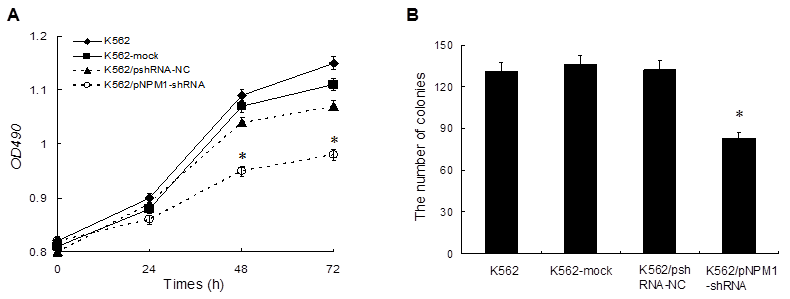
Effects of NPM1 gene silencing on the proliferation of K562 cells. A, Cells proliferation was determined by MTT assay. At 0 h, 24 h, 48 h, and 72 h, the value of OD in the four groups was measured by absorbance at 490 nm. Treatment of K562 cells with pNPM1-shRNA resulted in growth inhibition in a time-dependent manner. Data were shown as means ± SD of triplicate experiments. *p<0.05. B, Proliferation potential of cells was determined by colony formation assay. The number of colonies in K562/pNPM1-shRNA cells was significantly reduced compared with the controls. Data were shown as mean ± SD of triplicate experiments, *p<0.05.
Figure 3
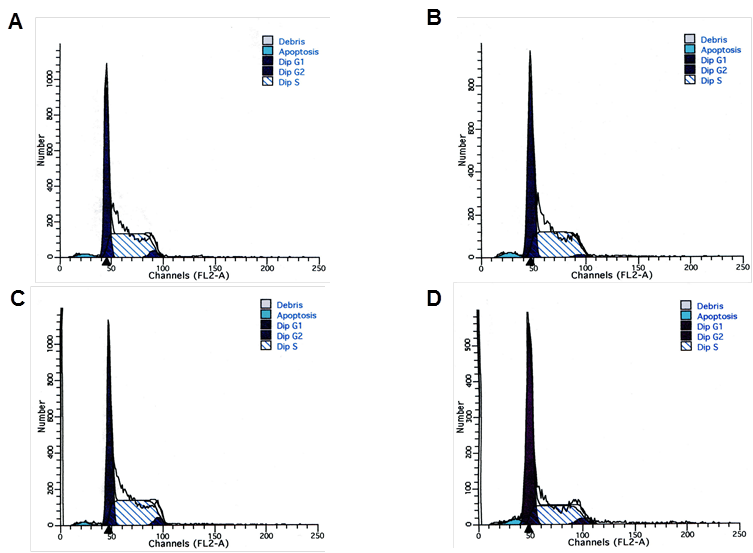
Effects of NPM1 gene silencing on cell cycle of K562 cells. The distribution of cell cycle in each group was analysis by Flow cytometry. A, K562 cells; B, K562-mock cells; C, K562/pshRNA-NC cells; D, K562/pNPM1-shRNA cells. A representative example of the experimental results is shown.
Table 1
Cell cycle distribution of transfected K562 cells
| Transfection group | G1 (%) | S (%) | G2 (%) |
|---|---|---|---|
| K562 | 44.75±3.97 | 52.86±4.63 | 2.40±1.14 |
| K562-mock | 44.19±2.27 | 51.30±2.63 | 4.48±0.90 |
| K562/pshRNA-NC | 47.21±2.72 | 48.58±2.84 | 4.23±1.20 |
| K562/pNPM1-shRNA | 57.51±1.67* | 36.87±2.88* | 5.62±2.14 |
Data representd mean ± SD of triplicate experiments, * p<0.05, as compared with K562, K562-mock and K562/pshRNA-NC.
Figure 4
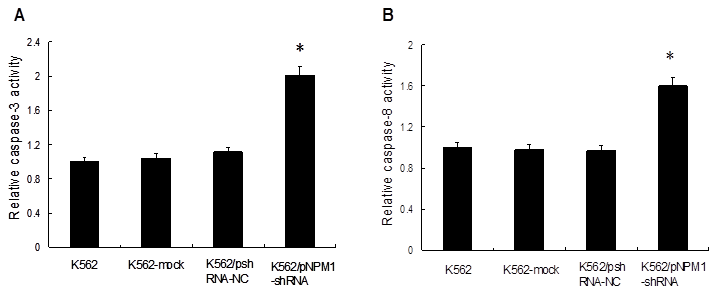
Effect of NPM1 gene silencing on the apoptosis of K562 cells. Caspases activities in the cells were measured by spectrophotometry. The relative activities of caspases in K562 cells were set as 1, and the values of other groups were standardized against it. Both caspase-3 and caspase-8 activities were significantly increased in K562/pNPM1-shRNA cells. Data were shown as mean ± SD of triplicate experiments, *p<0.05. A, Relative activity of caspase-3; B, Relative activity of caspase-8.
3.4 Down-regulation of NPM1 increased K562 cells apoptosis
To investigate the effect of NPM1 gene silencing on the apoptosis of K562 cells, the activities of caspase-3 and caspase-8 were detected. The results showed that, compared with the controls, the relative activity of caspase-3 increased by (2.01±0.04) fold in the cells of K562/pNPM1-shRNA group (p<0.05, Figure 4A), and the relative activity of caspase-8 increased by (1.60±0.07) fold (p<0.05, Figure 4B). However, no significantly change of caspase-3 or caspase-8 activity was observed in the controls (p>0.05, Figure 4A, B). These findings suggested that NPM1 silencing could increase the relative activities of caspases.
4. Discussions
NPM1, a multifunctional nucleolar protein, is found to be more abundant in tumor and aberrant of NPM1 has been proved to involve in the pathogenesis of diverse human malignancies. NPM1 may function as an oncogene by promoting aberrant cell growth and by inhibiting programmed cell death [23]. However, few of studies reported the oncogenic potential of the overexpression NPM1 in hematopoietic malignancies. Here, we identify that reduction of NPM1 by RNA interference can inhibit leukemic cells proliferation and induce cellular apoptosis.
In our study, RNA interference, a popular reverse genetic tool which inhibits gene expression through sequence-specific degradation of target mRNA was used. In order to avoid non-specific or off-target effects, three pairs of hairpin oligonucleotide sequences for NPM1 had been designed and synthesized, and the interference efficiency of each one had been detected in the pre-test (data were not shown). At last, the most effective one was selected for the further research. In this present study, the interference vector pNPM1-shRNA efficiently reduced NPM1 expression at both mRNA and protein levels. Since a nonspecific shRNA was also transfected as negative control, reduction of NPM1 was due to specific silent effect of pNPM1-shRNA. Our results showed a similar inhibit efficiency with Li et al [22], who silenced the NPM1 with the same target region in FA-associated leukemic cell line UoC-M1.
After confirming reduction of NPM1 in the K562/pNPM1-shRNA cells, the biological characteristics in vitro of leukemic cells were explored. MTT assays showed that the cells growth of K562/pNPM1-shRNA was significantly inhibited in a time-dependent manner. Meanwhile, the number of colonies in the K562/pNPM1-shRNA cells was significantly reduced compared with the controls. These findings indicated that reduction of NPM1 accounted for the inhibition of leukemia cells proliferation. NPM1 is reported to support cell growth based on its participation in synthesizing of ribosome and process the pre-rRNA molecule in the Internal transcribed spacer (ITS2) sequence [24], while knock-down of the NPM1 gene in murine cells leads to an alteration in their ribosome profile [5]. It indicates that down-regulation of NPM1 may impair its function on ribosome's synthesis, which may lead to cells growth block. In addition, we found the reduction of NPM1 significantly increased the K562/pNPM1-shRNA cells in G1 phase. Some studies reported that down-regulation of NPM1 mRNA by an antisense construct resulted in the delay of cells entering mitosis [25]. NPM1 expresses highly during S and G2 phases and is duplicates concomitantly with the initiation of DNA synthesis [23], which suggested that down-regulation of NPM1 may loss its duplicate function then delay the entry of cells into mitosis. As a result, down-regulation of NPM1 may block the cell cycle progression.
Besides cells proliferation potential, we detected the effect of NPM1 gene silencing on the apoptosis of the K562 cells. Caspase-3 plays a crucial role in apoptotic pathways and its expression is proportional to the cells apoptosis. Our results demonstrated that the relative activity of caspase-3 in K562/pNPM1-shRNA cells was significantly increased when compared with the controls, which indicated that reduction of NPM1 could induce apoptosis occurrence. In addition, we also detected the activity of caspase-8. Caspase-8 is often the apical caspase activated in the death receptor systems which serves to eliminate unwanted hematopoietic cells through apoptosis. Some studies have indicated that interferon regulatory factor 1 (IRF-1) could activate caspase-8 to induce apoptosis [26]. It also has been reported that overexpression of NPM1 suffered from IRF-1 inactivation and resist cell apoptosis [23]. In our study, the relative activity of caspase-8 in K562/pNPM1-shRNA cells was significantly increased, which indicated that induction of cellular apoptosis by silencing of NPM1 gene was associated with the change of caspase-8 activity. However, it is important to further study the exact molecular mechanisms of NPM1 on the apoptosis of leukemia cells.
In conclusion, our results demonstrated that inhibition of NPM1 decreased leukemia cells growth and induced cellular apoptosis. This new information casts a light on the oncogenic potential of the NPM1 overexpression in leukemia. However, to clarify the role of NPM1 in regulation of hematological malignancies, the effect of NPM1 overexpression on the cell biological behaviors still be investigated in the primary samples.
Acknowledgements
This project was supported by a grant from National Natural Science Foundation of China (No. 30872418) and Natural Science Foundation Project of CQ CSTC (No. 2010BB5363).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Chan WY, Liu QR, Borjigin J. et al. Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry. 1989;28:1033-9
2. Okuwaki M. The structure and functions of NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein. J Biochem. 2008;143:441-8
3. Sirri V, Roussel P, Gendron MC. et al. Amount of the two major Ag-NOR proteins, nucleolin and protein B23, is cell-cycle dependent. Cytometry. 1997;28:147-56
4. Wu MH, Yung BY. UV stimulation of nucleophosmin/B23 expression is an immediate-early gene response induced by damaged DNA. J Biol Chem. 2002;277:48234-40
5. Grisendi S, Bernardi R, Rossi M. et al. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147-53
6. Colombo E, Bonetti P, Lazzerini Denchi E. et al. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol Cell Biol. 2005;25:8874-86
7. Shields LB, Gerçel-Taylor C, Yashar CM. et al. Induction of immune responses to ovarian tumor antigens by multiparity. J Soc Gynecol Investig. 1997;4:298-304
8. Nozawa Y, Van Belzen N, Van der Made AC. et al. Expression of nucleophosmin/B23 in normal and neoplastic colorectal mucosa. J Pathol. 1996;178:48-52
9. Tanaka M, Sasaki H, Kino I. et al. Genes preferentially expressed in embryo stomach are predominantly expressed in gastric cancer. Cancer Res. 1992;52:3372-7
10. Subong EN, Shue MJ, Epstein JI. et al. Monoclonal antibody to prostate cancer nuclear matrix protein (PRO:4-216) recognizes nucleophosmin/B23. Prostate. 1999;39:298-304
11. Pianta A, Puppin C, Franzoni A. et al. Nucleophosmin is overexpressed in thyroid tumors. Biochem Biophys Res Commun. 2010;397:499-504
12. Feuerstein N, Chan PK, Mond JJ. The nuclear matrix protein, numatrin (B23), is associated with growth factor-induced mitogenesis in Swiss 3T3 fibroblasts and with T lymphocyte proliferation stimulated by lectins and anti-T cell antigen receptor antibody. J Cell Biol. 1988;107:1629-42
13. Kondo T, Minamino N, Nagamura-Inoue T. et al. Identification and characterization of nucleophosmin/B23/numatrin which binds the anti-oncogenic transcription factor IRF-1 and manifests oncogenic activity. Oncogene. 1997;15:1275-81
14. Morris SW, Xue L, Ma Z. et al. Alk+ CD30+ lymphomas: a distinct molecular genetic subtype of non-Hodgkin's lymphoma. Br J Haematol. 2001;113:275-95
15. Pandolfi PP. PML, PLZF and NPM genes in the molecular pathogenesis of acute promyelocytic leukemia. Haematologica. 1996;81:472-82
16. Yoneda-Kato N, Look AT, Kirstein MN. et al. The t (3; 5) (q25.1; q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. 1996;12:265-75
17. Hsu CY, Yung BY. Over-expression of nucleophosmin/B23 decreases the susceptibility of human leukemia HL-60 cells to retinoic acid-induced differentiation and apoptosis. Int J Cancer. 2000;88:392-400
18. Hsu CY, Yung BY. Involvement of nucleophosmin/B23 in TPA-induced megakaryocytic differentiation of K562 cells. Br J Cancer. 2003;89:1320-26
19. Liu WH, Hsu CY, Yung BY. Nucleophosmin/B23 regulates the susceptibility of human leukemia HL-60 cells to sodium butyrate-induced apoptosis and inhibition of telomerase activity. Int J Cancer. 1999;83:765-71
20. Liu WH, Yung BY. Mortalization of human promyelocytic leukemia HL-60 cells to be more susceptible to sodium butyrate-induced apoptosis and inhibition of telomerase activity by down-regulation of nucleophosmin/B23. Oncogene. 1998;17:3055-64
21. Bolli N, De Marco MF, Martelli MP. et al. A dose-dependent tug of war involving the NPM1 leukaemic mutant, nucleophosmin, and ARF. Leukemia. 2009;23:501-9
22. Li J, Zhang X, Sejas DP. et al. Negative regulation of p53 by nucleophosmin antagonizes stress-induced apoptosis in human normal and malignant hematopoietic cells. Leuk Res. 2005;29:1415-23
23. S. Grisendi, C. Mecucci, B. Falini, et al. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493-505
24. Herrera JE, Savkur R, Olson MO. The ribonuclease activity of nucleolar protein B23. Nucleic Acids Res. 1995;23:3974-79
25. Jiang PS, Yung BY. Down-regulation of nucleophosmin/B23 mRNA delays the entry of cells into mitosis. Biochem Biophys Res Commun. 1999;257:865-70
26. Bouker KB, Skaar TC, Riggins RB. et al. Interferon regulatory factor-1 (IRF-1) exhibits tumor suppressor activities in breast cancer associated with caspase activation and induction of apoptosis. Carcinogenesis. 2005;26:1527-35
Author contact
![]() Corresponding author: Ling Zhang, Department of Laboratory Medicine, Chongqing Medical University, 1#, Yixueyuan Road, Chongqing, 400016, China. Tel: +86 023-68485223, Fax: +86 023-68485005; Email: lingzhangedu.cn
Corresponding author: Ling Zhang, Department of Laboratory Medicine, Chongqing Medical University, 1#, Yixueyuan Road, Chongqing, 400016, China. Tel: +86 023-68485223, Fax: +86 023-68485005; Email: lingzhangedu.cn
Citation styles
APA
Qin, F.X., Shao, H.Y., Chen, X.C., Tan, S., Zhang, H.J., Miao, Z.Y., Wang, L., Hui-Chen, Zhang, L. (2011). Knockdown of NPM1 by RNA Interference Inhibits Cells Proliferation and Induces Apoptosis in Leukemic Cell Line. International Journal of Medical Sciences, 8(4), 287-294. https://doi.org/10.7150/ijms.8.287.
ACS
Qin, F.X.; Shao, H.Y.; Chen, X.C.; Tan, S.; Zhang, H.J.; Miao, Z.Y.; Wang, L.; Hui-Chen; Zhang, L. Knockdown of NPM1 by RNA Interference Inhibits Cells Proliferation and Induces Apoptosis in Leukemic Cell Line. Int. J. Med. Sci. 2011, 8 (4), 287-294. DOI: 10.7150/ijms.8.287.
NLM
Qin FX, Shao HY, Chen XC, Tan S, Zhang HJ, Miao ZY, Wang L, Hui-Chen, Zhang L. Knockdown of NPM1 by RNA Interference Inhibits Cells Proliferation and Induces Apoptosis in Leukemic Cell Line. Int J Med Sci 2011; 8(4):287-294. doi:10.7150/ijms.8.287. https://www.medsci.org/v08p0287.htm
CSE
Qin FX, Shao HY, Chen XC, Tan S, Zhang HJ, Miao ZY, Wang L, Hui-Chen, Zhang L. 2011. Knockdown of NPM1 by RNA Interference Inhibits Cells Proliferation and Induces Apoptosis in Leukemic Cell Line. Int J Med Sci. 8(4):287-294.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.