International Journal of Medical Sciences 3.2
Impact Factor
ISSN: 1449-1907
Impact Factor
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2011; 8(2):88-96. doi:10.7150/ijms.8.88 This issue Cite
Research Paper
Parvovirus B19 Nonstructural Protein-Induced Damage of Cellular DNA and Resultant Apoptosis
Brian D. Poole1,2, Violetta Kivovich1,3,4,5, Leona Gilbert4,5, Stanley J. Naides1, 5,6 ![]()
1. Huck Institute for Life Sciences, Pennsylvania State University College of Medicine/Milton S. Hershey Medical Center, Hershey, PA, USA
2. Current: Department of Microbiology and Molecular Biology, Brigham Young University, Provo, UT, USA
3. MD/PhD Program, Pennsylvania State University College of Medicine/Milton S. Hershey Medical Center, Hershey, PA, USA
4. Chemical Biology Division, Department of Biological and Environmental Science, University of Jyväskylä, Jyväskylä, Finland
5. Division of Rheumatology, Department of Medicine, Pennsylvania State University College of Medicine/Milton S. Hershey Medical Center, Hershey, PA, USA
6. Current: Quest Diagnostics Nichols Institute, San Juan Capistrano, CA, USA
Received 2010-12-2; Accepted 2011-1-13; Published 2011-1-15
Citation:
Poole BD, Kivovich V, Gilbert L, Naides SJ. Parvovirus B19 Nonstructural Protein-Induced Damage of Cellular DNA and Resultant Apoptosis. Int J Med Sci 2011; 8(2):88-96. doi:10.7150/ijms.8.88. https://www.medsci.org/v08p0088.htm
Other stylesAbstract
Parvovirus B19 is a widespread virus with diverse clinical presentations. The viral nonstructural protein, NS1, binds to and cleaves the viral genome, and induces apoptosis when transfected into nonpermissive cells, such as hepatocytes. We hypothesized that the cytotoxicity of NS1 in such cells results from chromosomal DNA damage caused by the DNA-nicking and DNA-attaching activities of NS1. Upon testing this hypothesis, we found that NS1 covalently binds to cellular DNA and is modified by PARP, an enzyme involved in repairing single-stranded DNA nicks. We furthermore discovered that the DNA nick repair pathway initiated by poly(ADPribose)polymerase and the DNA repair pathways initiated by ATM/ATR are necessary for efficient apoptosis resulting from NS1 expression.
Keywords: Parvovirus B19, DNA damage and repair, fulminant liver failure, apoptosis, autoantibody, systemic lupus erythematosus
Introduction
Parvovirus B19 (B19) is a common virus with multiple clinical presentations. Infection in children is typically seen as erythema infectiosum, or fifth disease (1), while adults often experience arthropathy lasting up to several months (2). Autoantibodies are often found subsequent to B19 infection, and are associated with arthropathy (3-5). In patients with chronic hemolytic anemias, such as sickle cell disease or hereditary spherocytosis, the destruction of the erythroid precursor pool by B19 leads to aplastic crisis (6). B19 infection is implicated in hepatitis non-A-E acute fulminant liver failure (7-16). Although these are the best-described clinical illnesses caused by B19, the virus has been implicated in a wide spectrum of other illnesses (17).
B19 infects a variety of cell types, but predominantly replicates in erythroid precursors (18). Infection of other cell types results in a limited, non-replicative state with overexpression of the viral nonstructural protein, NS1, and little expression of genes for the structural proteins VP1 and VP2 (19). Previous work in our laboratory showed that B19 is capable of infecting liver cells, and that the resulting restricted infection induces apoptosis, most likely through the action of NS1 (19, 20). NS1 is cytotoxic when transfected into erythroid cells (21), COS-7 cells (22) and liver-derived cells (20). In cell types which are non-productive for viral infection, NS1-induced apoptosis proceeds in a caspase 9-dependent manner, indicative of internal apoptotic stimuli (20, 22).
The NS1 protein of parvovirus B19 exhibits multiple functions, with NTP binding, helicase, nickase, and transcription factor activities (23-25). Because of these DNA-modifying activities, we hypothesized that NS1 induces apoptosis by damaging cellular DNA. Apoptosis resulting from DNA damage would be consistent with the caspase-9-dependent apoptotic pathway (20, 22).
This hypothesis is supported by the action of NS1 proteins from similar parvoviruses. The nonstructural proteins from the parvoviruses minute virus of mouse (MVM) and H-1 parvovirus also utilize helicase and DNA binding activities to fulfill their functions in viral replication (26-29). NS1 from MVM binds covalently to the viral genome as part of the replication process (29, 30). In addition, NS1 from MVM and H-1 parvovirus colocalizes with the cellular DNA repair machinery (31-33). Covalent attachment to cellular DNA would cause a significant lesion, as would the introduction of multiple single-strand breaks. DNA damage due to the actions of NS1 would be expected to result in apoptosis in a portion of infected cells.
This study utilized cloned NS1 under the control of an inducible promoter to examine the mechanisms of NS1-induced apoptosis. The NS1 DNA sequence was fused to that of green fluorescent protein (GFP) (http://tools.invitrogen.com/content/sfs/vectors/pindsp1gfp.pdf) to allow visualization and purification of NS1 (GFP/NS1). Cellular expression of this vector has previously been shown to induce apoptosis in the same manner as infection with natural B19, while a mutant of NS1 with the NTP binding region deleted induced significantly less apoptosis (20). The GFP/NS1 vector was utilized in this study to investigate the role of the DNA-damaging activities of NS1 in NS1-induced apoptosis.
There are several mechanisms through which NS1 could cause DNA damage resulting in apoptosis. We hypothesized that NS1 could covalently attach to chromosomal DNA, in much the same way that the nonstructural proteins of MVM and H-1 parvovirus attach to the viral genome. Covalent attachment of NS1 to cellular DNA was investigated in this study using denaturing SDS-PAGE and autoradiography. Attachment of NS1 to DNA would be expected to initiate the DNA repair pathways that sense distortions in the DNA helix. These pathways were examined by inhibition of the key proteins ataxia telangiectasia related (ATR) and ataxia telangiectasia-mutated (ATM). The DNA-nicking activity that NS1 uses to separate viral genomes would be expected to activate the single-strand break DNA repair pathway if applied to host cell DNA. This pathway was investigated by studying the activity of Poly(ADP-ribose)Polymerase (PARP), the protein which detects nicks in DNA and activates the repair process.
Both the nick repair and ATR/ATM-mediated bulky adduct repair pathways can result in apoptosis if the damage is severe. Damage of chromosomal DNA by parvoviral proteins has not been directly demonstrated, except in the case of specific integration of AAV. We present here evidence suggesting that NS1 is attached to DNA in a covalent manner, and that both DNA-helix distorting and single strand nick forms of DNA damage are important pathways to apoptosis upon expression of NS1.
Materials and Methods
Transfection
A GFP/NS1 expression vector under the control of the ecdysone response element previously constructed in our laboratory was utilized for these experiments as previously described (20). Briefly, HepG2 cells were grown on glass coverslips in hepatocyte wash medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum. Either GFP/NS1 or the parental vector, pIND(GFP)SP1 (Invitrogen) was cotransfected with the ecdysone receptor plasmid pVGRXR (Invitrogen) into the cells using Lipofectamine (Invitrogen) and PLUS reagent (Invitrogen). Expression of GFP/NS1 or GFP was induced by the addition of 10 µg/ml ponasterone A (Invitrogen). Protein expression was monitored by fluorescence microscopy.
Immunoprecipitation and chromatin immunoprecipitation
Cells expressing either GFP/NS1 or GFP were lysed with 1% SDS in TE buffer. The lysate was centrifuged through a Qiashredder (Qiagen, Valencia CA) to shear DNA. Lysates were then mixed with 2 ml of 1% triton-X 100 (Fisher, Hampton, NH) in PBS containing protease inhibitors (Sigma protease inhibitor cocktail, Sigma-Aldrich, St. Louis, MO). 25 μl of anti-GFP polyclonal antibody (Rockland, Gilbertsville, PA) were added and the mixture was allowed to bind for 14 hours at 4oC in an end-over end rotator. Immune complexes were bound to protein G-agarose beads (Pierce, Rockford, IL) for three hours at 4oC. Immunoprecipitates were washed 5x with 1% triton-X 100 in PBS, and once with PBS alone, and boiled for 5 minutes under reducing conditions in 1% SDS, 4M urea and 0.7 M 2-mercaptoethanol. Immunoprecipitates were electrophoresed on a 7.5-14% polyacrylamide gel and used for autoradiography and Western blotting.
For chromatin immunoprecipitation, 107 cells were cotransfected with pVGRXR and either GFP/NS1 or pIND(GFP)SP1. Protein expression was induced with ponasterone A 24 hours post-transfection and the cellular DNA was metabolically labeled with 10 µCui α32P thymidine triphosphate (Perkin Elmer) in supplemented hepatocyte wash medium (Invitrogen). After immunoprecipitation, one aliquot of each immunoprecipitate was treated with 10 units DNase (Roche) for 1 hour at 37oC. After SDS-PAGE, proteins were transferred to nitrocellulose and used to expose Kodak MS film to obtain an autoradiograph image. GFP antibodies were then used to identify the location of the transgenic protein by Western Blotting.
Western blotting
HepG2 cells were lysed in 1% (w/v) SDS, 4M urea and 0.7M 2-mercaptoethanol. Lysates were electrophoresed through 7.5-14% acrylamide gels (BioRad, Hercules, CA). Proteins were transferred to nitrocellulose membranes and bound with anti-GFP polyclonal rabbit antiserum (Invitrogen) or poly(ADP ribose) (PAR) monoclonal antibody (Pharmingen, San Diego, CA) at 1:5000 dilution. Species-specific secondary antibodies (Amersham, Piscataway, NJ) were used for detection with ECL+ chemiluminescence (Amersham).
Detection of apoptosis
Transfected HepG2 cells were grown on glass coverslips and stained with annexin-V-Alexa fluor 594 (Molecular Probes, Eugene, OR) as previously described (19, 20). Transfected cells were identified by green fluorescence and examined for apoptosis using a 528-553 nm excitation filter and a 600-660 nm barrier filter to allow for detection of the red-fluorescing apoptosis marker. Apoptotic cells also exhibited condensed nuclei when stained with Hoescht 33342 (Molecular Probes).
Treatment with pharmacologic agents
Transfected HepG2 cells were pretreated with 8 to 14 mM caffeine (Sigma) for 3 hours before induction of protein expression. Caffeine was maintained on the cells during expression of GFP or GFP/NS1. PARP was inhibited by incubating transfected cells with 5-aminoisoquinolinone (Calbiochem) at 250, 25, 2.5, and 0.25 μM concentrations 3 hours prior to transgene induction. The inhibitor was maintained on the cells throughout the experiment.
Statistical Analysis
The student's T test (2-tailed) was used to evaluate significant differences in the experiments involving inhibition of apoptosis, and the Pearson's correlation test was used to determine whether inhibition was dose-dependent. P values of less than 0.05 were considered significant.
Results
Covalent attachment of NS1 to chromosomal DNA
Association of NS1 with chromosomal DNA was detected using chromatin immunoprecipitation. HepG2 cells were transfected with the GFP/NS1 expression vector or GFP vector alone and metabolically labeled with 32P-thymidine triphosphate during the protein expression phase of the experiment. Labeled DNA was detected by autoradiography and proteins were detected by Western blotting. Radioactivity was found in specific bands that perfectly overlapped with bands formed by GFP/NS1, but not GFP, as revealed by Western blotting (Figure 1). The colocalization of the radioactive signal with NS1 shows that DNA is bound to NS1 in the lysate. The harsh denaturing methods used both in the immunoprecipitation and in the preparation of the samples for SDS-PAGE strongly suggest that DNA could not have been present with the NS1 fusion protein unless covalently linked. Treatment of the immunoprecipitate with DNase before SDS-PAGE decreased the radiographic signal by 63%, indicating that the radioactive label is DNA, and not from another source such as phosphorylation of NS1 (Figure 1).
Involvement of the DNA damage repair pathway
DNA damage normally blocks progression through the cell cycle and, when severe, causes apoptosis through the intrinsic or mitochondrial pathway. Caffeine uncouples DNA damage from cell cycle progression and apoptosis, primarily through the inhibition of the DNA damage sensing protein ATM and ATR, (34, 35). The involvement of the DNA damage repair pathway in NS1-induced apoptosis was examined in GFP/NS1-transfected cells by treating the cells with caffeine. Incubation of GFP/NS1-transfected cells with caffeine inhibited apoptosis in a dose-dependent manner, reducing the percentage of apoptotic cells by nearly 70% at a concentration of 14 mM (Figure 2).
Figure 1
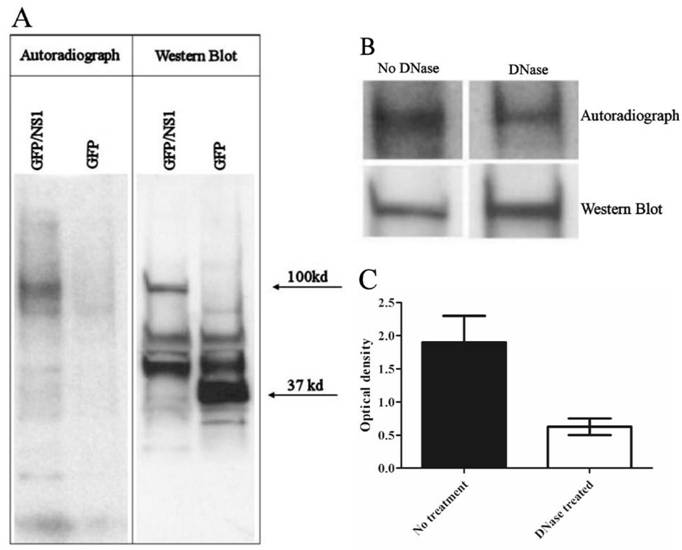
DNA is covalently bound to NS1 protein. A. Autoradiography of GFP-immunoprecipitated 32P labeled cells shows radioactive DNA colocalizing with GFP/NS1 (100 kd), but not GFP alone (37 kd), after boiling in SDS with urea and β-mercaptoethanol. GFP and GFP/NS1 were detected by western blot. The blot shown is representative of 4 independent experiments. B. Incubation of immunoprecipitates with DNase before the denaturation step abrogates the radiographic signal by 63% (N=3 experiments, error bars indicate the range).
Figure 2
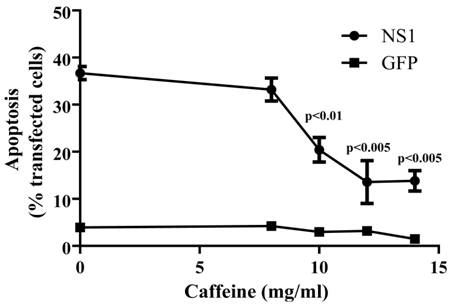
The ATM/ATR-mediated DNA repair pathways are necessary for efficient NS1-induced apoptosis. A. Caffeine treatment of GFP/NS1-transfected HepG2 cells led to a decrease in apoptosis of up to 63%, indicating the necessity for ATR-dependent activity in apoptosis. The decrease in apoptotic caffeine-treated cells compared to cells without caffeine treatment was significant by the student's t test for the three concentrations. Pearson correlation analysis comparing caffeine dose to apoptosis showed that the inhibition was dose-dependent (p<0.041). The data were derived from 3 independent experiments. Error bars indicate the standard error of the mean.
No difference was observed in apoptosis between the GFP-transfected cells and the untransfected cells upon treatment with caffeine. The decrease in apoptosis upon treatment with caffeine supports the finding that NS1 induces apoptosis through DNA damage that alters the chromatin structure.
Involvement of the DNA nick repair pathway
Although the ATM/ATR-dependent DNA repair pathway is important in optimal NS1-induced apoptosis, NS1 may also activate other DNA damage repair pathways that can lead to apoptosis. Single-strand nicks in genomic DNA would be expected to activate PARP and the nick repair pathway. Activated PARP transfers Poly(ADP ribose) (PAR) to neighboring proteins in response to DNA damage (36-38). As a method of investigating the involvement of PARP activation in NS1-induced apoptosis, the NS1 fusion protein was examined for the presence of activated PAR moieties, which would indicate the presence of NS1 in a DNA lesion that was sufficient to activate PARP, as well as demonstrating that the two molecules, NS1 and PARP were in physical contact. GFP/NS1 or GFP alone were immunoprecipitated from transfected cells, and western blotting was performed using an anti-PAR antibody. GFP/NS1 was poly(ADP ribose)ylated, while GFP was not (Figure 3A). Poly(ADP ribose)ylation of NS1 shows that NS1 exists in the cell in contact with activated PARP, and hence, in the presence of sufficient DNA nicks to activate this repair pathway. To study the importance of the PARP-initiated DNA repair pathway in NS1-induced apoptosis, the cell-permeable PARP inhibitor 5-aminoisoquinolinone (5AIQ) was added to GFP/NS1-transfected HepG2 cells. Inhibition of PARP significantly (p<0.003) reduced apoptosis in these cells compared to treatment with DMSO alone (Figure 3B). Inhibition of apoptosis was maximal at 57% at a concentration of 25 µM. This finding demonstrates that PARP activation, and therefore the PARP-induced DNA repair pathway, is an important mechanism of NS1-induced apoptosis.
Figure 3
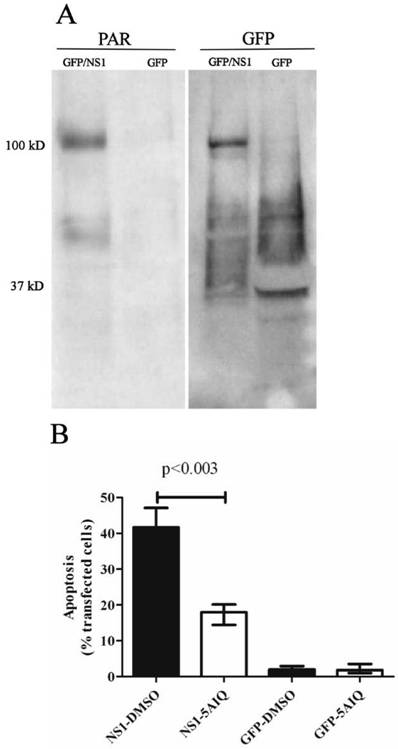
PARP is active and necessary for efficient apoptosis in NS1-transfected cells. A. Immunoprecipitated GFP/NS1 protein was poly(ADP ribose)ylated, as shown by a band at 100 kd on the blot probed with anti-poly ADP ribose (PAR, left blot) antibodies. The blots were stripped and reprobed with anti-GFP (right), showing that PAR colocalizes with the GFP/NS1 fusion protein. GFP alone is not ribosylated, as evidenced by the lack of a PAR band corresponding to GFP at 37 kD. Blots shown are representative of three independent experiments. B. PARP activity is necessary for optimal NS1-induced apoptosis. Addition of the PARP inhibitor 5-aminoisoquinolinone (5AIQ) to GFP/NS1 expressing HepG2 cells reduced apoptosis by 57% (p<0.003). Addition of 5-aminoisoquinolinone had no effect on the GFP transfected cells. N=3, error bars indicate the range of values.
Discussion
This work identifies several lines of evidence indicating that NS1 damages cellular DNA, and that this damage leads to apoptosis. Upon detection of DNA damage, DNA damage response proteins inhibit the cell cycle and are capable of inducing apoptosis if the DNA lesion is not repaired. Several of these repair pathways involve the DNA damage sensing kinases ATR and ATM. Upon activation, ATR and ATM phosphorylate a variety of substrates, including CHK-1, p53, and p73, each of which further transduces signals that result in DNA repair or apoptosis (39, 40). Blockage of the cell cycle has been noted in B19 and other parvovirus infected cells (21, 33, 41, 42), and p53 was implicated in NS1-induced apoptosis of COS-7 cells (22). These earlier findings suggest that NS1 may induce these DNA repair mechanisms. The experiments in this study are consistent with ATR/ATM-mediated DNA repair being important for parvovirus B19 NS1 protein-induced apoptosis. Inhibition of ATR and ATM with caffeine (34) substantially decreased the amount of apoptosis observed in the NS1-expressing cells. Although there are limitations inherent in these methods, the results presented are suggestive of DNA damage as a cause of NS1-induced apoptosis. ATM principally binds to free DNA ends or DNA strand breaks (43), while ATR recognizes single-stranded regions of DNA common to multiple types of DNA lesions and that are often caused by collapsed replication forks (44). NS1 could easily cause double strand breaks through the simple mechanism of nicking both DNA strands a short distance apart. Nicking and binding to the DNA end would not only create broken strands, but adducts that would likely interrupt replication and activate ATR-dependent DNA damage repair and apoptosis.
The pathway responsible for the repair of single-strand nicks in DNA is also important for NS1-induced apoptosis. This pathway is mediated through PARP. Upon binding DNA nicks, PARP transfers poly(ADP ribose) (PAR) chains to many of the surrounding proteins, leading to DNA repair and a decrease in the ATP levels of the cell (37, 38). If the damage to the DNA is extensive, both the adduct repair and nick repair pathways may result in apoptosis (37, 38, 45-49). Activation of PARP has been demonstrated to induce apoptosis in neuronal cells, to interfere with the electron potential of the mitochondria, and to be required for the translocation of apoptosis inducing factor from the mitochondria to the nucleus (36-38, 45).
The finding that NS1 is directly (ADP ribose)ylated in transfected cells provides direct evidence for an interaction between NS1 and PARP. Since PARP is active at the site of single strand DNA breaks, and single-strand nicking is a known activity of NS1 protein, it is likely that NS1 is nicking the cellular DNA, thereby inducing DNA damage. This damage leads to apoptosis, as shown by decreased apoptosis after PARP activity was inhibited with 5-aminoisoquinolinone.
The importance of DNA damage to NS1-induced apoptosis is shown by the abrogation of apoptosis in the presence of inhibitors of DNA damage recognition enzymes. Both the single strand nick repair pathway, mediated by PARP, and the helix-distorting damage repair pathways, mediated by ATM or ATR, are necessary for optimal induction of apoptosis induced by NS1 expression, since inhibition of these pathways significantly decreases apoptosis in transfected cells. It is likely that NS1 additionally uses mechanisms other than the DNA damage repair pathways studied here to induce apoptosis, given that some level of apoptosis persists even under maximal inhibitory conditions. However, the degree of inhibition seen upon administration of these agents indicates that DNA damage is the primary mechanism for NS1-induced apoptosis in these cells. It is likely that NS1 causes cell death by at least two mechanisms, primarily DNA damage-induced apoptosis in non-permissive cells and through other mechanisms in permissive cells.
These dual pathways correlate with differential viral expression in these cell types. In permissive cells, all B19 genes are transcribed, while in nonpermissive cells NS1 expression predominates (19, 50). NS1 expression leads to DNA damage in nonpermissive cells, where NS1 is over expressed, while the productive infection in permissive cells may lead to cell death by other mechanisms. One such method of inducing cell death is through TNF-α induced signaling (51). TNF-α was not involved in NS1-induced apoptosis of transfected HepG2 cells studied in our previous work (20). Other mechanisms that are responsible for cell death in permissive parvovirus infection include MVM NS1 interactions with Casein Kinase II, resulting in cytoskeletal rearrangement and cell death (52, 53), or the generation of reactive oxygen species in response to DNA damage induced by H1 parvovirus NS1 protein (54). Such mechanisms may contribute albeit to a lesser degree than direct DNA damage to the apoptosis seen in these cells. The relatively small number of cells killed by NS1 expression or B19 infection (approximately 1/3-1/2) can be explained if those cells that die are those that are both replicating and unable to adequately repair their DNA, while cells that can repair the damage survive. If NS1 were directly acting on apoptosis-inducing factors, increased cytotoxicity would be anticipated.
Single strand nicks are the type of DNA damage that would be expected from a parvovirus nonstructural protein that functions to separate the DNA replicative form into individual genomes. The process of nicking by NS1 in MVM is highly regulated, depending on both cellular factors, such as RPA, and recognition by NS1 of a specific DNA sequence (30, 55). The involvement of single strand nicks in B19 NS1-induced apoptosis of hepatocytes suggests that either the DNA-nicking activity by NS1 protein of B19 is not as highly regulated as in MVM, and so is able to proceed on cellular DNA, or that there are sites suitable for NS1 nicking on the chromosomes. Future studies analyzing the site at which NS1 binds to the chromatin should demonstrate the NS1 binding sequence.
An appealing possibility suggested by the finding of NS1-DNA attachment concerns the development of autoantibodies after B19 infection. B19 infection often results in the production of anti-DNA autoantibodies (3, 4). A viral protein, such as NS1, with a covalently linked DNA strand could serve as a hapten-carrier system. In this model, autoreactive B cells recognizing DNA in a DNA-protein complex could internalize the complex, and present peptides derived from NS1 to virus-specific T cells. NS1-specific T cells, generated in the normal immune response to B19 (56), could then activate the DNA-reactive B-cells that present NS1 peptides on their MHC molecules, leading to the development of autoantibodies. DNA and histones do in fact become immunogenic when bound by the large T antigen of polyomaviruses (57-60), a similar situation to what we envision with B19. Antigen presenting cell (APC) uptake of apoptotic bodies or immune complexes containing NS1- modified DNA would allow APC presentation of NS1 peptides on their MHC and activation of NS1-specific T cells. Parvovirus B19 may be an example of a virus that naturally induces anti-DNA antibodies in this manner (Figure 4).
This testable model for the generation of autoantibodies in response to B19 infection is attractive in that it assumes the utilization of T cells that are specific for viral NS1 protein, avoiding the need for autoreactive T cells, which tend to be tightly regulated. T cells specific for NS1 that are activated in the immune response against B19 would be sufficient to activate B cells to initiate the production of autoantibodies. Clinically, it is interesting when considering this model that patients with high titers of anti-NS1 antibodies also are more likely to develop arthropathy, a condition which may be a result of autoantibody production (5, 61-63). In addition to NS1, it is possible that autoimmune responses may be generated to other nuclear proteins associated with nucleosomal DNA or NS1.
Figure 4
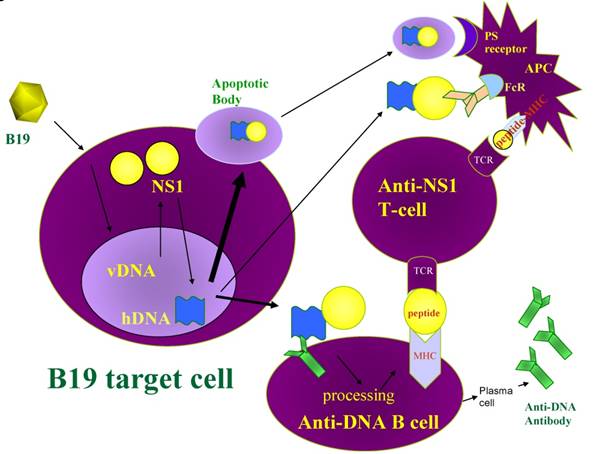
Model for B19 NS1 induction of anti-DNA antibodies. B19 induced apoptosis generates nucleosomes and apoptotic bodies containing NS1 modified DNA. Anergized anti-DNA B cells take up NS1 modified nucleosomal DNA through their anti-DNA immunoglobulin surface receptor and present NS1 peptides in the context of MHC to NS1-specific T cells. The NS1 specific T cells are activated by APC that express NS1 peptides in the context of surface MHC after uptake of apoptotic bodies or immune complexes containing NS1-modified DNA The NS1-specific T cells provide the helper signal required, in addition to the DNA signal, for the anergized B cell to break tolerance. vDNA, viral DNA; hDNA, human DNA; TCR, T cell receptor; PS receptor, phosphatidylserine receptor; FcR, Fc receptor.
Acknowledgements
We would like to thank Jing Zhou and Amy Grote for technical assistance. We would also like to thank Prof. Matti Vuento for his continuing support in this project. This work was supported in part by the Arthritis Foundation, Central Pennsylvania Chapter and the H. Thomas and Dorothy Willits Hallowell Endowment.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Anderson MJ, Jones SE, Fisher-Hoch SP, Lewis E, Hall SM, Bartlett CL. et al. Human parvovirus, the cause of erythema infectiosum (fifth disease)? Lancet. 1983;1(8338):1378
2. Naides SJ, Scharosch LL, Foto F, Howard EJ. Rheumatologic manifestations of human parvovirus B19 infection in adults. Initial two-year clinical experience. Arthritis Rheum. 1990;33(9):1297-309
3. Lunardi C, Tiso M, Borgato L, Nanni L, Millo R, De Sandre G. et al. Chronic parvovirus B19 infection induces the production of anti-virus antibodies with autoantigen binding properties. Eur J Immunol. 1998;28(3):936-48
4. Hansen KE, Arnason J, Bridges AJ. Autoantibodies and common viral illnesses. Semin Arthritis Rheum. 1998;27(5):263-71
5. Jones LP, Erdman DD, Anderson LJ. Prevalence of antibodies to human parvovirus B19 nonstructural protein in persons with various clinical outcomes following B19 infection. J Infect Dis. 1999;180(2):500-4
6. Cubel RC, Valadao MC, Pereira WV, Magalhaes MC, Nascimento JP. Aplastic crisis due to human parvovirus B19 infection in hereditary hemolytic anaemia. Rev Inst Med Trop Sao Paulo. 1992;34(5):479-82
7. Dutta U, Mittal S, Ratho RK, Das A. Acute liver failure and severe hemophagocytosis secondary to parvovirus B19 infection. Indian J Gastroenterol. 2005;24(3):118-9
8. Ho JK, Tha SP, Coupland R, Dalal BI, Bowie WR, Sreenivasan GM. et al. Parvovirus B19 in an immunocompetent adult patient with acute liver failure: an underdiagnosed cause of acute non-A-E viral hepatitis. Can J Gastroenterol. 2005;19(3):161-2
9. Karetnyi YV, Beck PR, Markin RS, Langnas AN, Naides SJ. Human parvovirus B19 infection in acute fulminant liver failure. Arch Virol. 1999;144(9):1713-24
10. Kishore J, Sen M. Parvovirus B19-induced thrombocytopenia and anemia in a child with fatal fulminant hepatic failure coinfected with hepatitis A and E viruses. J Trop Pediatr. 2009Oct;55(5):335-7
11. Langnas AN, Markin RS, Cattral MS, Naides SJ. Parvovirus B19 as a possible causative agent of fulminant liver failure and associated aplastic anemia. Hepatology. 1995;22(6):1661-5
12. Lee WM, Brown KE, Young NS, Dawson GJ, Schlauder GG, Gutierrez RA. et al. Brief report: no evidence for parvovirus B19 or hepatitis E virus as a cause of acute liver failure. Dig Dis Sci. 2006;51(10):1712-5
13. Ozcay F, Bikmaz YE, Canan O, Ozbek N. Hepatitis A and parvovirus B19 infections in an infant with fulminant hepatic failure. Turk J Gastroenterol. 2006;17(2):148-50
14. Pardi DS, Romero Y, Mertz LE, Douglas DD. Hepatitis-associated aplastic anemia and acute parvovirus B19 infection: a report of two cases and a review of the literature. Am J Gastroenterol. 1998;93(3):468-70
15. So K, Macquillan G, Garas G, Delriviere L, Mitchell A, Speers D. et al. Urgent liver transplantation for acute liver failure due to parvovirus B19 infection complicated by primary Epstein-Barr virus and cytomegalovirus infections and aplastic anaemia. Intern Med J. 2007;37(3):192-5
16. Tung J, Hadzic N, Layton M, Baker AJ, Dhawan A, Rela M. et al. Bone marrow failure in children with acute liver failure. J Pediatr Gastroenterol Nutr. 2000;31(5):557-61
17. Bultmann BD, Klingel K, Sotlar K, Bock CT, Kandolf R. Parvovirus B19: a pathogen responsible for more than hematologic disorders. Virchows Arch. 2003;442(1):8-17
18. Srivastava A, Lu L. Replication of B19 parvovirus in highly enriched hematopoietic progenitor cells from normal human bone marrow. J Virol. 1988;62(8):3059-63
19. Poole BD, Karetnyi YV, Naides SJ. Parvovirus B19-induced apoptosis of hepatocytes. J Virol. 2004;78(14):7775-83
20. Poole BD, Zhou J, Grote A, Schiffenbauer A, Naides SJ. Apoptosis of liver-derived cells induced by parvovirus B19 nonstructural protein. J Virol. 2006;80(8):4114-21
21. Moffatt S, Yaegashi N, Tada K, Tanaka N, Sugamura K. Human parvovirus B19 nonstructural (NS1) protein induces apoptosis in erythroid lineage cells. J Virol. 1998;72(4):3018-28
22. Hsu TC, Wu WJ, Chen MC, Tsay GJ. Human parvovirus B19 non-structural protein (NS1) induces apoptosis through mitochondria cell death pathway in COS-7 cells. Scand J Infect Dis. 2004;36(8):570-7
23. Fu Y, Ishii KK, Munakata Y, Saitoh T, Kaku M, Sasaki T. Regulation of tumor necrosis factor alpha promoter by human parvovirus B19 NS1 through activation of AP-1 and AP-2. J Virol. 2002;76(11):5395-403
24. Zhi N, Mills IP, Lu J, Wong S, Filippone C, Brown KE. Molecular and functional analyses of a human parvovirus B19 infectious clone demonstrates essential roles for NS1, VP1, and the 11-kilodalton protein in virus replication and infectivity. J Virol. 2006;80(12):5941-50
25. Raab U, Beckenlehner K, Lowin T, Niller HH, Doyle S, Modrow S. NS1 protein of parvovirus B19 interacts directly with DNA sequences of the p6 promoter and with the cellular transcription factors Sp1/Sp3. Virology. 2002;293(1):86-93
26. Jindal HK, Yong CB, Wilson GM, Tam P, Astell CR. Mutations in the NTP-binding motif of minute virus of mice (MVM) NS-1 protein uncouple ATPase and DNA helicase functions. J Biol Chem. 1994;269(5):3283-9
27. Nuesch JP, Christensen J, Rommelaere J. Initiation of minute virus of mice DNA replication is regulated at the level of origin unwinding by atypical protein kinase C phosphorylation of NS1. J Virol. 2001;75(13):5730-9
28. Nuesch JP, Corbau R, Tattersall P, Rommelaere J. Biochemical activities of minute virus of mice nonstructural protein NS1 are modulated In vitro by the phosphorylation state of the polypeptide. J Virol. 1998;72(10):8002-12
29. Nuesch JP, Cotmore SF, Tattersall P. Sequence motifs in the replicator protein of parvovirus MVM essential for nicking and covalent attachment to the viral origin: identification of the linking tyrosine. Virology. 1995;209(1):122-35
30. Christensen J, Tattersall P. Parvovirus initiator protein NS1 and RPA coordinate replication fork progression in a reconstituted DNA replication system. J Virol. 2002;76(13):6518-31
31. Young PJ, Jensen KT, Burger LR, Pintel DJ, Lorson CL. Minute virus of mice NS1 interacts with the SMN protein, and they colocalize in novel nuclear bodies induced by parvovirus infection. J Virol. 2002;76(8):3892-904
32. Cziepluch C, Lampel S, Grewenig A, Grund C, Lichter P, Rommelaere J. H-1 parvovirus-associated replication bodies: a distinct virus-induced nuclear structure. J Virol. 2000;74(10):4807-15
33. Bashir T, Rommelaere J, Cziepluch C. In vivo accumulation of cyclin A and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J Virol. 2001;75(9):4394-8
34. Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM. et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59(17):4375-82
35. Blasina A, Price BD, Turenne GA, McGowan CH. Caffeine inhibits the checkpoint kinase ATM. Curr Biol. 1999;9(19):1135-8
36. Nicoletti VG, Stella AM. Role of PARP under stress conditions: cell death or protection? Neurochem Res. 2003;28(2):187-94
37. Smulson ME, Simbulan-Rosenthal CM, Boulares AH, Yakovlev A, Stoica B, Iyer S. et al. Roles of poly(ADP-ribosyl)ation and PARP in apoptosis, DNA repair, genomic stability and functions of p53 and E2F-1. Adv Enzyme Regul. 2000;40:183-215
38. Wang H, Shimoji M, Yu SW, Dawson TM, Dawson VL. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Ann N Y Acad Sci. 2003;991:132-9
39. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3(3):155-68
40. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265-79
41. Op De Beeck A, Caillet-Fauquet P. The NS1 protein of the autonomous parvovirus minute virus of mice blocks cellular DNA replication: a consequence of lesions to the chromatin? J Virol. 1997;71(7):5323-9
42. Op De Beeck A, Sobczak-Thepot J, Sirma H, Bourgain F, Brechot C, Caillet-Fauquet P. NS1- and minute virus of mice-induced cell cycle arrest: involvement of p53 and p21(cip1). J Virol. 2001;75(22):11071-8
43. Lavin MF, Khanna KK. ATM: the protein encoded by the gene mutated in the radiosensitive syndrome ataxia-telangiectasia. Int J Radiat Biol. 1999;75(10):1201-14
44. Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci. 2010 [Epub ahead of print]
45. Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ. et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259-63
46. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616-27
47. Kulkarni A, Das KC. Differential roles of ATR and ATM in p53, Chk1, and histone H2AX phosphorylation in response to hyperoxia: ATR-dependent ATM activation. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L998-L1006
48. Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12(9):440-50
49. Sinclair A, Yarranton S, Schelcher C. DNA-damage response pathways triggered by viral replication. Expert Rev Mol Med. 2006;8(5):1-11
50. Bonvicini F, Filippone C, Manaresi E, Zerbini M, Musiani M, Gallinella G. Functional analysis and quantitative determination of the expression profile of human parvovirus B19. Virology. 2008;381(2):168-77
51. Sol N, Le Junter J, Vassias I, Freyssinier JM, Thomas A, Prigent AF. et al. Possible interactions between the NS-1 protein and tumor necrosis factor alpha pathways in erythroid cell apoptosis induced by human parvovirus B19. J Virol. 1999;73(10):8762-70
52. Nuesch JP, Rommelaere J. NS1 interaction with CKII alpha: novel protein complex mediating parvovirus-induced cytotoxicity. J Virol. 2006;80(10):4729-39
53. Nuesch JP, Rommelaere J. A viral adaptor protein modulating casein kinase II activity induces cytopathic effects in permissive cells. Proc Natl Acad Sci U S A. 2007;104(30):12482-7
54. Hristov G, Kramer M, Li J, El-Andaloussi N, Mora R, Daeffler L. et al. Through its nonstructural protein NS1, parvovirus H-1 induces apoptosis via accumulation of reactive oxygen species. J Virol. 2010;84(12):5909-22
55. Christensen J, Cotmore SF, Tattersall P. Minute virus of mice initiator protein NS1 and a host KDWK family transcription factor must form a precise ternary complex with origin DNA for nicking to occur. J Virol. 2001;75(15):7009-17
56. Klenerman P, Tolfvenstam T, Price DA, Nixon DF, Broliden K, Oxenius A. T lymphocyte responses against human parvovirus B19: small virus, big response. Pathol Biol (Paris). 2002;50(5):317-25
57. Bredholt G, Olaussen E, Moens U, Rekvig OP. Linked production of antibodies to mammalian DNA and to human polyomavirus large T antigen: footprints of a common molecular and cellular process? Arthritis Rheum. 1999;42(12):2583-92
58. Rekvig OP, Moens U, Sundsfjord A, Bredholt G, Osei A, Haaheim H. et al. Experimental expression in mice and spontaneous expression in human SLE of polyomavirus T-antigen. A molecular basis for induction of antibodies to DNA and eukaryotic transcription factors. J Clin Invest. 1997;99(8):2045-54
59. Moens U, Seternes OM, Hey AW, Silsand Y, Traavik T, Johansen B. et al. In vivo expression of a single viral DNA-binding protein generates systemic lupus erythematosus-related autoimmunity to double-stranded DNA and histones. Proc Natl Acad Sci U S A. 1995;92(26):12393-7
60. Andreassen K, Moens U, Nossent H, Marion TN, Rekvig OP. Termination of human T cell tolerance to histones by presentation of histones and polyomavirus T antigen provided that T antigen is complexed with nucleosomes. Arthritis Rheum. 1999;42(11):2449-60
61. Zakrzewska K, Azzi A, De Biasi E, Radossi P, De Santis R, Davoli PG. et al. Persistence of parvovirus B19 DNA in synovium of patients with haemophilic arthritis. J Med Virol. 2001;65(2):402-7
62. Mitchell LA, Leong R, Rosenke KA. Lymphocyte recognition of human parvovirus B19 non-structural (NS1) protein: associations with occurrence of acute and chronic arthropathy? J Med Microbiol. 2001;50(7):627-35
63. Kerr JR, Cunniffe VS. Antibodies to parvovirus B19 non-structural protein are associated with chronic but not acute arthritis following B19 infection. Rheumatology (Oxford). 2000;39(8):903-8
Author contact
![]() Corresponding author: Stanley J. Naides, MD, Immunology, Quest Diagnostics Nichols Institute, 33608 Ortega Highway, San Juan Capistrano, CA 92690. Tel. 949 728-4578; fax 949 728-7852; email: stanley.j.naidescom
Corresponding author: Stanley J. Naides, MD, Immunology, Quest Diagnostics Nichols Institute, 33608 Ortega Highway, San Juan Capistrano, CA 92690. Tel. 949 728-4578; fax 949 728-7852; email: stanley.j.naidescom
Citation styles
APA
Poole, B.D., Kivovich, V., Gilbert, L., Naides, S.J. (2011). Parvovirus B19 Nonstructural Protein-Induced Damage of Cellular DNA and Resultant Apoptosis. International Journal of Medical Sciences, 8(2), 88-96. https://doi.org/10.7150/ijms.8.88.
ACS
Poole, B.D.; Kivovich, V.; Gilbert, L.; Naides, S.J. Parvovirus B19 Nonstructural Protein-Induced Damage of Cellular DNA and Resultant Apoptosis. Int. J. Med. Sci. 2011, 8 (2), 88-96. DOI: 10.7150/ijms.8.88.
NLM
Poole BD, Kivovich V, Gilbert L, Naides SJ. Parvovirus B19 Nonstructural Protein-Induced Damage of Cellular DNA and Resultant Apoptosis. Int J Med Sci 2011; 8(2):88-96. doi:10.7150/ijms.8.88. https://www.medsci.org/v08p0088.htm
CSE
Poole BD, Kivovich V, Gilbert L, Naides SJ. 2011. Parvovirus B19 Nonstructural Protein-Induced Damage of Cellular DNA and Resultant Apoptosis. Int J Med Sci. 8(2):88-96.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.