Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2010; 7(4):181-190. doi:10.7150/ijms.7.181 This issue Cite
Research Paper
ISOLATION OF CHLAMYDIA PNEUMONIAE FROM SERUM SAMPLES OF THE PATIENTS WITH ACUTE CORONARY SYNDROME
Ivan M Petyaev 1, Nayilia A Zigangirova 2, Alexey M Petyaev 3, Ulia P Pashko 2, Lubov V Didenko2, Elena U Morgunova 2, Yuriy K Bashmakov1 ![]()
1. Cambridge Theranostics Ltd, Babraham Research Campus, Babraham, Cambridge, CB2 4AT, United Kingdom
2. Gamaleya Institute for Epidemiology and Microbiology RAMS, 18 Gamaleya Str., Moscow 123098, Russia
3. Rostov-on-Don Medical University. Nahichevanskii 37, Rostov-on-Don, Russia
Received 2009-12-16; Accepted 2010-6-7; Published 2010-6-10
Abstract
BACKGROUND: Limited body of evidence suggests that lipopolysaccharide of C. pneumoniae as well as C. pneumoniae-specific immune complexes can be detected and isolated from human serum. The aim of this study was to investigate the presence of viable elementary bodies of C.pneumoniae in serum samples of patients with acute coronary syndrome and healthy volunteers.
MATERIAL AND METHODS: Serum specimens from 26 healthy volunteers and 56 patients with acute coronary syndrome were examined subsequently by serological (C.pneumoniae-specific IgA and IgG), PCR-based and bacteriological methods. Conventional, nested and TaqMan PCR were used to detect C.pneumoniae genetic markers (ompA and 16S rRNA) in DNA from serum specimens extracted with different methods. An alternative protocol which included culturing high-speed serum sediments in HL cells and further C.pneumoniae growth evaluation with immunofluorescence analysis and TaqMan PCR was established. Pellet fraction of PCR-positive serum specimens was also examined by immunoelectron microscopy.
RESULTS: Best efficiency of final PCR product recovery from serum specimens has been shown with specific C. pneumoniae primers using phenol-chloroform DNA extraction protocol. TaqMan PCR analysis revealed that human serum of patients with acute coronary syndrome may contain genetic markers of C. pneumoniae with bacterial load range from 200 to 2000 copies/ml serum. However, reliability and reproducibility of TaqMan PCR were poor for serum specimens with low bacterial copy number (<200 /ml). Combination of bacteriological, immunofluorescence and PCR- based protocols applied for the evaluating HL cells infected with serum sediments revealed that 21.0 % of the patients with acute coronary syndrome have viable forms C.pneumoniae in serum. The detection rate of C.pneumoniae in healthy volunteers was much lower (7.7%). Immunological profile of the patients did not match accurately C.pneumoniae detection rate in serum specimens. Elementary bodies of C.pneumoniae with typical ultrastructural characteristics were also identified in serum sediments using immunoelectron microscopy.
Conclusions: Viable forms C. pneumoniae with typical electron microscopic structure can be identified and isolated from serum specimens of the patients with acute coronary syndrome and some healthy volunteers. Increased detection rate of C. pneumoniae in serum among the patients with an acute coronary syndrome may contribute towards enhanced pro-inflammatory status in cardiovascular patients and development of secondary complications of atherosclerosis.
Keywords: Chlamydia pneumoniae, PCR, human serum, acute coronary syndrome, cultured cells
BACKGROUND
Despite unquestionable role of C. pneumoniae in pathogenesis of respiratory infections there are many questions about involvement of the pathogen in development other human diseases including atherosclerosis (1), multiple sclerosis (2,3), Alzheimer's disease (4), lymphogranuloma (5), reactive arthritis (6), Guillain-Barre syndrome (7). The progress in that field is substantially complicated by the lack of standardized criteria for laboratory diagnostics of chronic C. pneumoniae infection as well as contradictory information about distribution of the pathogen throughout of the tissues of human body.
Isolating and culturing of C. pneumoniae may represent significant challenge for non-specialized diagnostic labs. Several plasma serological markers have been recently proposed based on the results of proteomic analysis. In particular proteins encoded by Omp11, the PmpG family, IncA and by CpPLD are among promising candidates for immunological diagnostics of C. pneumoniae infection (8, 9). However, changed antigenic profile of C. pneumoniae during persistent colonization in human tissues (10, 11) undermines the diagnostic value of serological markers.
Among molecular diagnostic criteria used for detection of C. pneumoniae in human specimens are polymerase chain reaction (PCR), in-situ hybridization method and enzyme immunoassay protocols (12, 13). PCR-based approach usually targets parts of chlamydial genome, in particular genes encoding 16S rRNA, major outer membrane protein (OmpA), as well as Pst1 (13).
However poor reproducibility limits significantly the diagnostic importance of PCR and in-situ hybridization for non-respiratory specimens. Detection of chlamydial lipopolysaccharide in serum is claimed to improve reliability of molecular biology methods when used in addition to PCR and in situ hybridization protocols (12).
There are multiple reports validating the presence of C. pneumoniae in respiratory secretion fluid, nasal, tracheal and lung tissues of the patients with inflammatory lung disease (13, 14, 15). Moreover, C. pneumoniae can efficiently propagate in blood cells, in particular in mononuclear cells and lymphocytes (16,17,18). The presence of C. pneumoniae in the blood cells predetermines the possibility of pathogen dissemination from respiratory system to different organs and tissues. Besides respiratory organs C. pneumoniae can be detected in specimens from atherosclerotic plagues (1, 19), cerebrospinal fluid (2) and endothelium (20).
In the present paper we report, that viable elementary bodies of C. pneumoniae with typical electron microscopic structure can be isolated from the serum samples of the patients with acute coronary syndrome. Furthermore, using combination of bacteriological and PCR-based methods we show herein that patients with acute coronary syndrome have higher C. pneumoniae detection rate in serum as compared to healthy volunteers.
MATERIAL AND METHODS
Cell lines and bacterial strains
HL cells (Washington Research Foundation, Seattle, USA) as well as C. pneumoniae (strain Kajaani 6, K6) were kindly provided by Dr. P.Saikku (University of Oulu, Finland). HL cells were grown in RPMI 1640 supplemented with 10% FCS at 37° C in 5% CO2. C.pneumoniae was initially propagated in HL cells and elementary bodies (EB) were purified by Renografin gradient centrifugation as widely described (21, 22). EB of C. pneumoniae were used as a reference for genetic and electron microscopy analysis.
Patients and serum specimens
The study protocol was approved by the Rostov-on- Don Medical University Ethics Committee. All patients were informed about the purpose of the study and have given written consent regarding participation in the study. Initial observation has been done on the group of 18 patients with acute coronary syndrome (11 males and 7 females aged from 47 to 68). Once conditions for combined microbiologic and nucleic acid amplification protocol were established, 38 more patients with acute coronary syndrome (21 males and 17 females, aged from 42 to 71) and 26 healthy volunteers with no indication of cardiovascular disease were enrolled (major groups of the study). Blood samples were collected into plastic tubes, kept at 37° C for 20 minutes and centrifuged at 1000g, 4° C for 10 min. Resulting serum was immediately separated and stored at - 80° C until assayed.
C.pneumoniae-specific IgA and IgG antibodies were evaluated by using Chlamydia pneumoniae-IgG-ELISA medac plus and Chlamydia pneumoniae-IgA-ELISA plus commercial kits with highly purified C.pneumoniae specific antigen without LPS. (Medac, Hamburg, Germany).
Bacteriological assay
Tubes containing 3 ml of frozen serum samples were thawed on ice and subjected to the centrifugation on Beckman centrifuge AN (Beckman Coulter, Inc., USA) at 16000 g for 45 min at 4° C. Obtained sediments were gently resuspended with micropipette in 1.0 ml of RPMI 1640 with 5% FCS, amphothericine B (5 µg/ml) and gentamycin (4 µg/ml). Resulting suspension was transferred to subconfluent monolayer of HL cells grown in 24- well plate. After inoculation the plates were centrifuged at 1600g for 1 hour at 30° C and incubated for 2 h at 37° C in 5% C02. The medium was removed and replaced with fresh RPMI 1640 supplemented with 1 µg/ml) of cycloheximide and plates were cultivated for 72 hours at 37° C in 5% CO2.
A 24 well plate rather than 96 well plates was used in the study to avoid potential cross contamination. Each serum specimen inoculated into 24 well dish was followed by two wells filled with incubation medium alone. All manipulation with the plates were done without agitation. Positive control plates were set and examined by the end of each working day and were kept in separate incubator. Each plate examination procedure was followed by careful disinfection of the equipment. Positive findings were reconfirmed.
The plates were evaluated for chlamydial growth by immunofluorescence microscopy with a Chlamydia genus-specific antibody against LPS prior to quantitative TaqMan- PCR for 16S rRNA of C. pneumoniae. Each isolate was passaged up to 3 times.
Immunofluoresence staining
Infected HL monolayers grown on coverslips in 24-well plates were fixed with methanol. Permebialized cells were stained by direct immunofluoresence using FITC - conjugated monoclonal antibody against chlamydial lipopolysaccharide (NearMedic Plus, RF). Inclusion-containing cells were visualized using Nikon Eclipse 50i microscope fluorescence microscope at x1350 magnification.
DNA isolation
DNA isolation from whole serum
Briefly, 1.0 ml of whole serum was mixed with 0.5 ml of lysis buffer (0.2 M Tris-HCl buffer, pH 7,2 supplemented with 0.5 % SDS) with 0.25 mg/ ml proteinase K (Promega, USA) and incubated for 2 hours at 56° C. DNA from the resulting lysates was extracted using phenol-chloroform method as widely described (23) and precipitated with absolute ethanol. DNA pellet was finally resuspended in 25 µl of water. For comparison purpose bacterial DNA was extracted from the same volume of whole serum with QIAmp Blood Midi Kit (QIAGEN, Valencia, CA) according to the manual.
Bacterial DNA was also extracted from the bacterial particles trapped from the whole serum with protein A from of Staph. aureus, insoluble (Sigma P7155). 1.0 ml of whole serum was mixed with 0.15 ml of protein A and incubated for 1 hour at 37 °C with occasional gentle shaking. The mixture was centrifuged for 5 min at 5000 g and DNA was extracted from the resulting pellet using QIAamp DNA Blood Mini Kit (QIAGEN INC., Valencia, Calif.) according to the manual.
DNA isolation from infected HL cells
Cells were harvested from 24 well plates and resuspended in 200 µl of lysis buffer and DNA was extracted using QIAamp DNA Blood Mini Kit (QIAGEN INC., Valencia, Calif.) according to the manual.
DNA isolation from C.pneumoniae reference strain
DNA was extracted from 100 µl of C. pneumoniae purified EB using reagents and protocol from QIAmp Blood Mini Kit (QIAGEN Inc., Valencia, Calif.).
PCR
General Information
Numerous precautions were employed to ensure validity of PCR protocols, especially nested PCR. Different work areas/rooms, different sets of the pipets, barrier-filter tips and scrupulous cleaning/decontamination procedures were used. All samples were blinded for lab workers. Multiple controls were used for PCR reactions. DNA extracted from C. pneumoniae reference strain (low concentration) and/or DNA extracted from the serum sediment of two C.pneumoniae infected patients were used as positive control. Positive control specimens were selected using electron microscopy and serological assay. Serum specimens from serologically negative healthy volunteers with no C. pneumoniae EB detectable in serum sediments by electron microcopy were used as a negative control. Each PCR set was accompanied by a reaction mix with all PCR components except the target DNA. Positive findings were reconfirmed.
Conventional qualitative PCR
Briefly, 2 µl of DNA solution were transferred to the reaction mixture containing 1x PCR buffer (Silex, Moscow, RF) containing 10 mM Tris-HCl, pH 8,3 , 2.5 mM MgCl2, 200 µM of each dNTPs, 1 U Taq-DNA-polymerase, 15 pmol of each primer. Forty-five cycles of amplification were performed on a PCR Thermocycler Perkin Elmer. Each cycle consisted of denaturation step at 94°C for 45 sec, primer annealing at 63°C for 45 sec , primer extension at 72°C for 45 sec. Amplified product (10 µl) was visualized by electrophoresis in a 1.5% agarose gel with ethidium bromide. Extracted DNAs were analyzed by PCR with primers CPN90-CPN91 specific for C. pneumoniae 16S rRNA as described (24).
Nested PCR
To ensure the specificity of PCR analysis a protocol for nested PCR for OmpA of C. pneumoniae was employed. The outer (oCP1 - 5' TTACAAGCCTTGCCTGTAGG 3', oCP2 - 5' GCGATCCCAAATGTTTAAGGC 3') and nested (iCPC - 5' TTATTAATTGATGGTACAATA 3', iCPD - 5' ATCTACGGCAGTAGTATAGTT 3') primers were used as published (24).
2 µl of DNA was added to reaction mixture containing 1x PCR buffer (Silex, Moscow, RF) containing 10 mM Tris-HCl, pH 8,3 , 2.5 mM MgCl2, 15 pmol of each primer, 200 µM of each of dNTPs and 1 U of Taq polymerase. First run of amplification was conducted under cycling conditions consisting of an initial denaturation at 95°C for 5 min, followed by 45 cycles of denaturation at 95°C for 30 sec, annealing at 63°C for 30 sec, and extension for 30sec at 72°C. For the second round of PCR, 2 µl of the first-round product was mixed with 23 µl of amplification mixture containing primers for iCPC and iCPD and amplified using following cycling conditions: 35 cycles of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec, and extension for 30sec at 72°C. PCR products were visualized by agarose electrophoresis with ethidium bromide. Taq DNA polymerase and other reagents for nested PCR were from Promega (UK).
Quantitative TaqMan-PCR
For quantification purpose, Real-time PCR for 16S rRNA of C. pneumoniae was conducted. PCR primers and TaqMan probe for 16S rRNA (GenBank accession number AF131889) were designed using Primer Express Software (Applied Biosystems, Foster City, CA, USA) and synthesized by Syntol (Moscow, RF). Designed primers and TaqMan probe (forward primer СPN90, 5'-GGTCTCAACCCCATCCGTGTCGG-3'; reverse primer СPN91, 5'-TGCGGAAAGCTGTATTTCTACAGTT-3'; and TaqMan probe 557, 5'-TCCAGGTAAGGTCCTTCGCGTTGCATCG-3') generated a PCR product of predicted size (194 bp). The TaqMan probe was labelled at the 5' end with 6-carboxyfluorescein as the reporter dye and at the 3' end with 6-carboxytetramethylrhodamine as the quencher. An additional BLAST search analysis was conducted to unsure specificity of the primers and probe. Real-time PCR was performed with the iCycler IQ ystem (Biorad, USA). 2 µl of extracted DNA was analyzed with the PCR mixture in a total volume of 25 µl. The PCR mixture consisted of 10 mM Tris (pH 8.3), 50 mM KCl, 1,5 mM MgCl2, 200 µM of each dNTPs, 2,5 U of Termostar Taq DNA polymerase (Syntol, Moscow, RF); and 5pmol of both forward and reverse primers and 3,5 pmol probe. The real-time PCR run was 10 min at 95°C, and 50 repeats of 20 sec at 95°C and 50 sec at 62°C. All samples were analyzed in triplicates. A sample was considered positive if three of three assay results were positive in the triplicate test and if the average value for the PCR run was greater than or equal to 1.0.
Amounts of 16S rRNA are represented bellow in 16S rRNA genome equivalents per ml of serum. Calibrator standards were prepared using 194 bp 16S rRNA DNA fragment of C pneumoniae cloned into the pGEM-T plasmid vector (pVU56) using the TA cloning kit (Invitrogen, San Diego, CA) similarly to Broccolo F (25).
The cycle threshold (CT) values, defined as the number of cycles at which the fluorescence of the reporter dye first exceeds the calculated background level, were automatically estimated by the instrument for each reaction. CT values for serum samples were plotted against calibrator standards of cloned DNA fragment.
Electron Microscopy
Thawed serum samples (10 ml) were spun at 16000g for 60 min. Resulting pellets were analyzed by TaqMan PCR for C. pneumoniae 16S rRNA. Positive specimens were fixed for 4 hours in phosphate buffer (pH 7.8) containing 5% glutaraldehyde, post-fixed in 1% osmium tetroxide for 1 hour, dehydrated in ethanol and embedded in LR White resin (EMS, USA). Stained ultrathin sections (200-300Аº) were evaluated by electron microscopy using JEM-100B microscope (Japan Electron Optics Laboratory Co., Tokyo, Japan). Purified EB of C. pneumoniae reference strain were used as positive control for electron microscopy studies. PCR-negative sediments of serum obtained from healthy volunteers served as negative control.
Immunoelectron microscopy was performed in specimens fixed with 2% paraformaldehyde and 0.1 % glutaraldehyde in PBS (7.5) with further contrasting with 2% uranyl acetate. Acetone-dehydrated specimens were embedded into LR White Resin for ultrathin sectioning. The sections were blocked for 1 hour with 0.5% bovine serum albumin in PBS and incubated overnight with monoclonal antibody against chlamydial lipopolysaccharide (NearMedic Plus, RF). After washing in PBS sections were incubated for 2 hours with goat anti-mouse IgG conjugated with 10 nm colloid gold (Invitrogen, USA) and contrasted with uranyl acetate. Sections were examined with a Joel 100B (Japan) electron microscope. Control sections were incubated with normal mouse IgG.
RESULTS
Initial observation took place when we obtained sera from 18 patients with ACS and analyzed them for presence of C. pneumoniae specific IgG and IgA using ELISA Medac kit (Germany) as well as for presence of genomic determinants of C. pneumoniae. As can be seen from Table 1, 7 patients from the initial group were positive for C. pneumoniae-specific IgG, whereas 4 patients had diagnostically relevant levels of IgA. Simultaneous detection of increased titers of IgG and IgA was documented only in 4 patients. Surprisingly, when DNA specimens extracted from 1.0 ml of serum aliquots were analyzed for presence of 16S rRNA by conventional PCR, we have found that 5 patients with ACS were positive for the genetic marker of C. pneumoniae. Finally, just 3 patients (out of 18) had increased levels of two Ig isotypes and positive signal in conventional PCR for 16S rRNA. Such inconsistency between serologic and genetic markers of C. pneumoniae infection is well known and widely discussed (1). However, detectability of the genetic marker of C. pneumoniae in human serum appeared to be a reasonably intriguing finding. Therefore, we decided to optimize conventional PCR protocol for detection of the C. pneumoniae genetic markers in serum.
C.pneumoniae positivity status assessment using serological, RT- PCR and bacteriological analysis of serum specimens
| GROUPS | Total | Number of individuals Positive in: | ||||
|---|---|---|---|---|---|---|
| Serological assay | TaqMan PCR in serum | Bacteriological assay with further PCR validation | ||||
| IgA | IgG | |||||
| PRELIMINARY GROUP | Patients with ACS | 18 | 4 | 7 | 5 | 3 |
| MAYOR GROUPS | Healthy volunteers | 26 | 1 | 4 | - | 2 |
| Patients with ACS | 38 | 6 | 13 | - | 8 | |
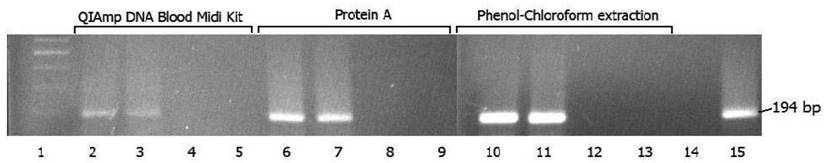
PCR-positive sera obtained from 2 randomly selected ACS patients were used for this purpose. As can be seen from Figure 1, there is an obvious increase in the final recovery of 194 bp PCR product (16S rRNA amplicon) when phenol-chloroform DNA extraction protocol has been used. Somehow QIAmp Midi Extraction kit (Qiagen) showed lower recovery rate of final PCR product which can be explained by lower efficiency of C. pneumoniae DNA extraction. Sufficient recovery of final PCR product has been also seen when protein A from Staph. aureus has been used for isolation of C pneumoniae from whole serum. This fact may suggest that extracted DNA originates rather from intact chlamydial particles opsonized by immunoglobulins, than remnants of C. pneumoniae circulating in the blood.
Recovery of PCR product in DNA samples isolated from serum specimens using QIAamp DNA blood midi kit, protein A and phenol-chloroform extraction method. 1 - molecular size standards; 2, 6 and 10 - PCR-positive serum from patient M; 3, 7 and 11 - PCR-positive serum from patient P; 4, 8 and 12 - PCR negative serum from patient S; 5, 9 and 13 - extraction control; 14 - negative control; 15 - positive control.
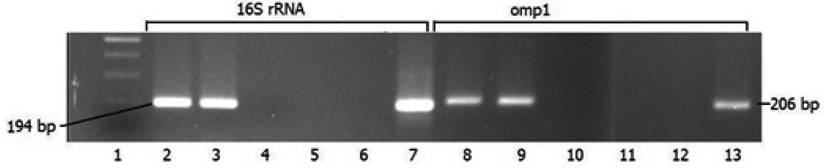
To confirm the results obtained with conventional PCR and insure its specificity we compared side-by-side two amplification reactions with phenol-chloroform extracted C. pneumoniae DNA. One has been conducted with protocol using primers specific for 16S rRNA, another one - with the primers for ompA in nested PCR format. As can be seen from Figure 2, the sensitivity of PCR reaction was similar regardless of the primer set used.
Recovery of PCR products in amplification reactions with different primers (chlamydial 16 S rRNA and omp1) using DNA extracted from human plasma by phenol-chloroform method. 1 - molecular size standards; 2, and 8 - PCR positive serum from patient M.; 3 and 9 - PCR positive serum from patient P.; 4 and 10 - PCR negative serum from patient S.; 5 and 11 - extraction control; 6 and 12 - negative controls; 7 and 13 - positive controls.
Next, we decided to employ TaqMan PCR protocol for quantification of C. pneumoniae DNA extracted from serum specimens with phenol-chloroform method. Standard curves were made using incremental dilutions of reference plasmid containing the primer-spanning region of C. pneumoniae 16S rRNA gene. These standard plasmid dilutions covered the range of plasmid concentration corresponding from 5 to 1,0 6 copies/µl. According to the results obtained from the initial group, 5 serum specimens obtained from the patients with ACS had a positive TaqMan PCR assay with variations in bacterial load from 200 to 2000 copies/ml of serum. However 2 serum specimens with lowest copy numbers (<300 copies/ml) had inconsistent PCR readings with CT values exceeding 30 cycles on two or more different attempts. Thus, TaqMan assay validates the presence of C.pneumoniae DNA in the serum samples of the patients with ACS. However it is clear that TaqMan PCR is associated with some sensitivity and reproducibility issues when C. pneumoniae is present in serum specimens at low copy number.
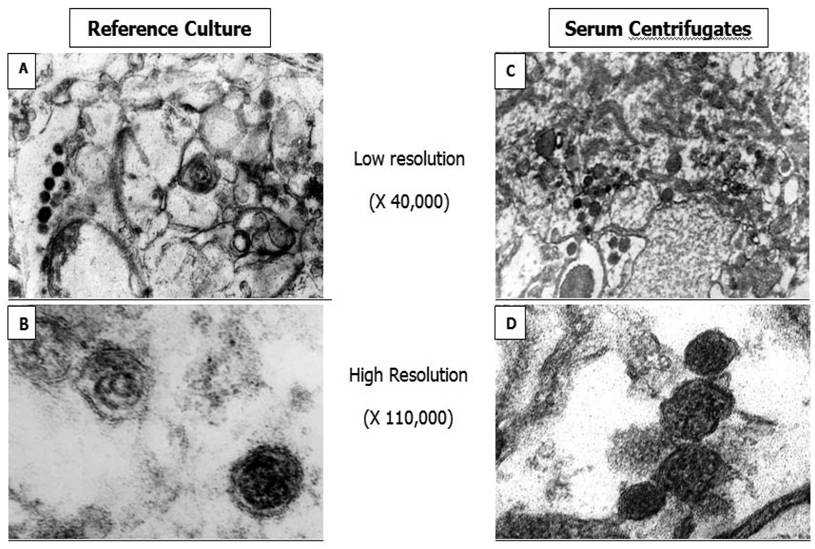
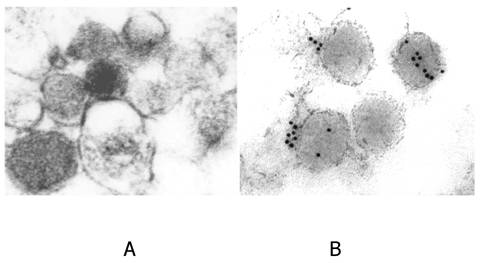
To confirm the presence of C pneumoniae in serum specimens obtained from the patients with ACS we also used ultrastructural analysis. 16000g serum sediments from two patients were analyzed first by TaqMan assay and further electron microcopy method. As can be seen from Figure 3, 16000 g pellet fraction contained visually intact pear shaped electron-dense structures approximately 0.3 microns in diameter similar to elementary bodies of C. pneumoniae reference strain. Both serum sediments were positive in TaqMan PCR assay as well. Therefore, electron microscopy analysis supports PCR data demonstrating the presence of C. pneumoniae in serum specimens of the patients with ACS. The identity of ultrastructures in serum sediments was confirmed by immune-gold labeling protocol (Figure 4). However obtained results do not answer a question about viability of C. pneumoniae particles present in the serum specimens.
Electron-microscopic images of C.pneumoniae elementary bodies obtained from HL cells infected with C. pneumoniae reference strain (A and B) and serum centrifugates (C and D).
Electron-microscopic images. Immunogold labeling of C.pneumoniae elementary bodies in serum sediments. A - preincubation with normal mouse IgG. B - preincubation with monoclonal antibody against chlamydial LPS.
To address this issue we decided to implement a combination of classical bacteriological protocol and nucleic acid amplification method in detection of C. pneumoniae in serum specimens of ACS patients. Statistically representative group of ACS patients (Table 1, clinical trial group) as well as age- and sex-matched control group were used for this purpose. According to the results diagnostically relevant levels of IgG for C. pneumoniae were found in 34.2% of the patients with ACS, whereas IgA positive were seen just in 19.7% of the ACS patients. Simultaneous detection of increased IgG and IgA took place just in 13.1 % of the ACS patients. Control group had a lower detection rate for both IgG and IgA (19.3% and 3,8% respectively).
16000 g pellet fraction of serum specimens obtained from 38 patients with ACS were resuspended and inoculated into cycloheximide-treated HL cells for further culture assay and immunofluroscence analysis with chlamydial LPS-specific antibody. A total of 8 specimens from ACS patients were positive for chlamydial growth with LPS-specific antibody revealing the presence of viable C. pneumoniae in serum. Positive immunofluorescence was usually seen within 72 hours after specimen inoculation. Inclusions varied in size and staining but were generally much smaller, than usually seen in case of C. pneumoniae infection in cultured cells and had reduced intensity of immunofluorescent staining (Figure 5) resembling those reported during persistent C pneumoniae infection in presence of IFN-γ (26).
Immunofluoresence analysis of HL monolayers after inoculation of serum sediments (A), and reference culture (B) of C. pneumoniae.

All 10 serum isolates survived at least 2 passages and were tested positive by 16S rRNA TaqMan-PCR assay, suggesting the identity of the isolates as C. pneumoniae. Just two serum specimen obtained from the control group had been confirmed to be positive for C.pneumoniae by culture assay and PCR analysis despite of lack diagnostically relevant titers of IgG and IgA. Only 4 patients with ACS were assessed positively by both bacteriological protocol and Medac IgG-IgA assay.
DISCUSSION
C. pneumoniae is an obligate intracellular respiratory pathogen which can be identified in different tissues and organs. Dissemination of the pathogen is believed to be mediated by peripheral blood mononuclear cells known to harbor viable chlamydial particles (27). It is possible to culture C. pneumoniae from monocytes of cardiovascular patients suggesting that the ability to form virulent elementary bodies is not lost within the mononuclear cell (28). However, the elementary bodies of C. pneumoniae have never been found in free circulation within the bloodstream or any other bodily fluid except cerebrospinal fluid (2).
The major conclusion from the results presented above is that viable and virulent forms of C. pneumoniae can be isolated from the human serum. Despite atypical visual appearance of the inclusion bodies, the isolates can infect, survive and multiply in the host cells accomplishing full cycle of chlamydial infection. Moreover, genetic markers of C. pneumoniae (16S rRNA and omp1) as well as ultrastructures identified by immunoelectron microscopy as elementary bodies of Chlamydia spp. can be detected and in the human serum. These results were supported by nested PCR protocol and TaqMan PCR assay. The later revealed that chlamydial load in serum specimens varies among the individuals in the range of 200-2000 copies/ml of serum. The performance of nucleic acid amplification protocols used in our studies was highly dependable on the method DNA extraction and bacterial load value in the serum specimens. In particular, regardless of targeted sequence (16S rRNA or omp1), best PCR performance has been achieved with DNA extracted with phenol-chloroform protocol, whereas TaqMan PCR sensitivity became unsatisfactory at low values of bacterial load. On the other hand, serum contains many PCR inhibitors (29) which undermines the usage of quantitative PCR in direct testing of serum specimens. Therefore, in the current attempt to evaluate the prevalence of C pneumoniae bacteremia in cardiovascular patients we used a combination of cell culture technique with further evaluation of isolates by PCR. That approach confirmed our preliminary results and allowed us to find, that 21% of the patients with ACS appear to be positive for presence of C. pneumoniae in serum specimens. Much lower detection rate has been seen in control group (7.6%). Unfortunately, detectability of C. pneumoniae in serum specimens is in controversial agreement with seropositivity rate in Medac IgG and IgA assay. Currently, IgA level is considered to be indicative of active pathogen due to the short life of IgA (30). Nevertheless even this parameter alone does not match accurately the status of exposure of the patients to C. pneumoniae measured by cell culture test with further PCR.
To our best knowledge, our manuscript is a first communication reporting the isolation of C. pneumoniae from serum specimens. However, there are some other communications supporting indirectly our finding. In particular, it is well known that human serum contains detectable amounts of chlamydial LPS and C. pneumoniae-specific immune complexes (12, 31). In our view, serum-associated LPS is likely to derive from partially destroyed cell walls and apparently from intact elementary bodies of C. pneumoniae.
However, we have to acknowledge that our results do not resolve any current problems in laboratory diagnostics of chlamydial infection. The protocol used in our study is hardly adjustable to routine work in regular diagnostic laboratory since it requires cultured cells and significant volumes of serum. A sensitive PCR protocol is urgently in need for quantification of chlamydial bacterial load in clinical specimens. Some other questions need to be addressed in future research. First of all, our results do not reveal the origin of chlamydial particles in human serum. It is possible, that they may originate from destructed mononuclear cells and/or macrophages residing in the atherosclerotic plaques. However regardless of the origin, we can claim that the presence of virulent chlamydial particles in serum is an apparent sign of C. pneumoniae circulation in the bloodstream and may represents a new potential mechanism of the pathogen generalization throughout the human body. At the same time, we realize disputable relevance of our results to pathogenesis and clinical manifestations of atherosclerosis. The finding of C. pneumoniae in serum of ACS patients does not establish causality for the pathogen in development of atherosclerosis. However, increased positivity rate for presence of C. pneumoniae in serum among ACS patients is very likely to contribute towards enhanced pro-inflammatory status in cardiovascular patients and development of secondary complications of atherosclerosis.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Shor A. Chlamydia atherosclerosis lesion, discovery, diagnosis and treatment. Springer-Verlag. 2007
2. Yi-Wei T, Subramaniam S, Haijing L. et al. Qualitative and Quantitative Detection of Chlamydophila pneumoniae DNA in Cerebrospinal Fluid from Multiple Sclerosis Patients and Controls. PLoS ONE. 2009;4(4):e5200
3. Stratton CW, Wheldon DB. Multiple sclerosis: an infectious syndrome involving Chlamydophila pneumoniae. Trends Microbiol. 2006Nov;14(11):474-9
4. Stallings TL. Association of Alzheimer's disease and Chlamydophila pneumoniae. J Infect. 2008Jun;56(6):423-31
5. Shen D, Yuen HK, Galita DA. et al. Detection of Chlamydia pneumoniae in a bilateral orbital mucosa-associated lymphoid tissue lymphoma. American Journal Of Ophthalmology. 2006Jun;141(6):1162-3
6. Carter JD, Gérard HC, Espinoza LR. et al. Chlamydiae as etiologic agents in chronic undifferentiated spondylarthritis. Arthritis Rheum. 2009May;60(5):1311-6
7. Haidl S, Ivarsson S, Bjerre I. et al. Guillain-Barre syndrome after Chlamydia pneumoniae infection. N Engl J Med. 1992;326:576-7
8. Park SH, Kwon SJ, Lee SJ. et al. Identification of Immunogenic Antigen Candidate for Chlamydophila pneumoniae Diagnosis. J Proteome Res. 2009Jun;8(6):2933-43
9. Mancini F, Savarino A, Losardo M. et al. Characterization of the serological response to phospholipase D protein of Chlamydophila pneumoniae in patients with acute coronary syndromes. Microbes Infect. 2009Mar;11(3):367-73
10. Maäurer AP, Mehlitz A, Mollenkopf HJ. et al. Gene Expression Profiles of Chlamydophila pneumoniae during the Developmental Cycle and Iron Depletion-Mediated Persistence. PLoS Pathog. 2007;3(6):e83
11. Polkinghorne A, Hogan RJ, Vaughan L. et al. Differential expression of chlamydial signal transduction genes in normal and interferon gamma-induced persistent Chlamydophila pneumoniae infections. Microbes Infect. 2009;8:61-72
12. Lajunen T, Vikatmaa P, Ikonen T. et al. Comparison of polymerase chain reaction methods, in situ hybridization, and enzyme immunoassay for detection of Chlamydia pneumoniae in atherosclerotic carotid plaques. Diagn Microbiol Infect Dis. 2008Jun;61(2):156-64
13. Loens K, Beck T, Ursi D. et al. Development of Real-Time Multiplex Nucleic Acid Sequence-Based Amplification for Detection of Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella spp in Respiratory Specimens. J Clin Microbiol. 2008;46(1):185-91
14. Teig N, Anders A, Schmidt C. et al. Chlamydophila pneumoniae and Mycoplasma pneumoniae in respiratory specimens of children with chronic lung diseases. Thorax. 2005;60(11):962-6
15. Brandén E, Gnarpe J. et al. Detection of Chlamydia pneumoniae on cytospin preparations from bronchoalveolar lavage in COPD patients and in lung tissue from advanced emphysema. Int J COPD. 2007Dec;2(4):643-50
16. Airenne S, Surcel HM, Alakarppa H. et al. Chlamydia pneumoniae infection in human monocytes. Infect Immun. 1999;67:1445-9
17. Müller J, Holm C, Nyvad O. et al. Repetitive measurements of Chlamydia pneumoniae DNA in peripheral blood mononuclear cells in healthy control subjects and dialysis patients. Scand J Infect Dis. 2004;36(10):718-23
18. Anil P, Terttu T, Mirja P. et al. Chlamydia pneumoniae DNA is present in peripheral blood mononuclear cells during acute coronary syndrome and correlates with chlamydial lipopolysaccharide levels in serum. Scand J Infect Dis. 2009;41(3):201-5
19. Iriz E, Cirak MY, Engin ED. et al. Effects of atypical pneumonia agents on progression of atherosclerosis and acute coronary syndrome. Acta Cardiol. 2007Dec;62(6):593-8
20. Schmeck B, Beermann W, N'Guessan PD. et al. Simvastatin reduces Chlamydophila pneumoniae-mediated histone modifications and gene expression in cultured human endothelial cells. Circ Res. 2008Apr;102(8):888-95
21. Galdwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia Trachomatis. Infect. Immun. 1981;31(3):1161-76
22. Maniatis T, Fritsch E. F, Sambrook J. Molecular cloning: a laboratory manual. New York, USA: Cold Spring Harbor. 1989
23. Madico G, Quinn TC, Boman J. et al. Touchdown enzyme time release-PCR for detection and identification of Chlamydia trachomatis, C. pneumoniae, and C. psittaci using the 16S and 16S-23S spacer rRNA genes. J Clin Microbiol. 2000Mar;38(3):1085-93
24. Boulos M, Hans B, Marjolein K. et al. Is the Perceived Association between Chlamydia pneumoniae and Vascular Diseases Biased by Methodology. J Clin Microbiol. 2004 Sep: 3937-41
25. Broccolo F, Locatelli G, Sarmati L. et al. Calibrated Real-Time PCR Assay for Quantitation of Human Herpesvirus 8 DNA in Biological Fluids. J Clin Microbiol. 2002Dec;40(12):4652-8
26. Pantoja LG, Miller RD, Ramirez JA. et al. Characterization of Chlamydia pneumoniae persistence in HEp-2 cells treated with gamma interferon. Infect Immun. 2001Dec;69(12):7927-32
27. Rupp J, Koch M, van Zandbergen G. et al. Transmission of Chlamydia pneumoniae infection from blood monocytes to vascular cells in a novel transendothelial migration model. FEMS Microbiol Lett. 2005Jan15;242(2):203-8
28. Cirino F, Webley W.C, West C. et al. Detection of Chlamydia in the peripheral blood cells of normal donors using in vitro culture, immunofluorescence microscopy and flow cytometry technique. BMC Infect Dis. 2006;6:23-9
29. Mygind T, Birkelund S, Birkebaek NH. et al. Determination of PCR efficiency in chelex-100 purified clinical samples and comparison of real-time quantitative PCR and conventional PCR for detection of Chlamydia pneumoniae. BMC Microbiol. 2002Jul;2:17
30. Contini C, Seraceni S, Cultera R, Castellazzi M, Granieri E, Fainardi E. Chlamydophila pneumoniae infection and its role in neurological disorders. Interdisciplinary Perspectives on Infectious Diseases. 2010:273573
31. Parratt J, Tavendale R, O'Riordan J. et al. Chlamydia pneumoniae-specific serum immune complexes in patients with multiple sclerosis. Mult Scler. 2008Apr;14(3):292-9
Author contact
![]() Corresponding author: Dr Yuriy K Bashmakov, Cambridge Theranostics Ltd., Babraham Research Campus, Cambridge CB2 4AT, United Kingdom. Telephone: +44-797-1598348, Fax: +44-122-3240340
Corresponding author: Dr Yuriy K Bashmakov, Cambridge Theranostics Ltd., Babraham Research Campus, Cambridge CB2 4AT, United Kingdom. Telephone: +44-797-1598348, Fax: +44-122-3240340