International Journal of Medical Sciences
ISSN: 1449-1907
3.2
Impact Factor
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2010; 7(3):147-154. doi:10.7150/ijms.7.147 This issue Cite
Research Paper
NITRIC OXIDE (NO), CITRULLINE - NO CYCLE ENZYMES, GLUTAMINE SYNTHETASE AND OXIDATIVE STRESS IN ANOXIA (HYPOBARIC HYPOXIA) AND REPERFUSION IN RAT BRAIN
M. Swamy ![]() , Mohd Jamsani Mat Salleh, K. N .S. Sirajudeen, Wan Roslina Wan Yusof, G. Chandran
, Mohd Jamsani Mat Salleh, K. N .S. Sirajudeen, Wan Roslina Wan Yusof, G. Chandran
Department of Chemical Pathology, School of Medical Sciences, Health campus, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
Received 2009-12-30; Accepted 2010-5-26; Published 2010-5-31
Citation:
Swamy M, Salleh MJM, Sirajudeen KNS, Yusof WRW, Chandran G. NITRIC OXIDE (NO), CITRULLINE - NO CYCLE ENZYMES, GLUTAMINE SYNTHETASE AND OXIDATIVE STRESS IN ANOXIA (HYPOBARIC HYPOXIA) AND REPERFUSION IN RAT BRAIN. Int J Med Sci 2010; 7(3):147-154. doi:10.7150/ijms.7.147. https://www.medsci.org/v07p0147.htm
Other stylesAbstract
Nitric oxide is postulated to be involved in the pathophysiology of neurological disorders due to hypoxia/ anoxia in brain due to increased release of glutamate and activation of N-methyl-D-aspartate receptors. Reactive oxygen species have been implicated in pathophysiology of many neurological disorders and in brain function. To understand their role in anoxia (hypobaric hypoxia) and reperfusion (reoxygenation), the nitric oxide synthase, argininosuccinate synthetase, argininosuccinate lyase, glutamine synthetase and arginase activities along with the concentration of nitrate /nitrite, thiobarbituric acid reactive substances and total antioxidant status were estimated in cerebral cortex, cerebellum and brain stem of rats subjected to anoxia and reperfusion. The results of this study clearly demonstrated the increased production of nitric oxide by increased activity of nitric oxide synthase. The increased activities of argininosuccinate synthetase and argininosuccinate lyase suggest the increased and effective recycling of citrulline to arginine in anoxia, making nitric oxide production more effective and contributing to its toxic effects. The decreased activity of glutamine synthetase may favor the prolonged availability of glutamic acid causing excitotoxicity leading to neuronal damage in anoxia. The increased formation of thiobarbituric acid reactive substances and decreased total antioxidant status indicate the presence of oxidative stress in anoxia and reperfusion. The increased arginase and sustained decrease of GS activity in reperfusion group likely to be protective.
Keywords: Citrulline - Nitric oxide cycle, Nitric oxide, Anoxia, Hypobaric hypoxia, Reperfusion, Excitotoxicity, Glutamine synthetase, Thiobarbituricacid reactive substances, Total antioxidant status
Introduction
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system (CNS) (1). It has the potential to be involved in the pathogenesis of many CNS diseases either due to excessive release, reduced uptake or alteration of receptor function (2). Neuronal excitotoxicity usually refers to injury and death of neurons arising from prolonged exposure to glutamate and associated excessive influx of ions into the cell. The resulting calcium overload is particularly neurotoxic, leading to the activation of enzymes that degrade proteins, membranes and nucleic acids (3). Glutamate is released from damaged axons and glia under hypoxic/ischemic conditions (4) and glutamate receptor-mediated excitotoxicity has been described as a predominant mechanism of hypoxic injury to the developing cerebral white matter (5-8). In the CNS, the conversion of glutamate to glutamine by glutamine synthetase (GS; EC 6.3.1.2), that takes place within the astrocytes, represents a key mechanism in the regulation of excitatory neurotransmission under normal conditions as well as in injured brain (9). Thus GS is involved in modulation of the turnover of glutamate through the glutamate-glutamine cycle (10). Reactive oxygen species (ROS) are free radicals that are normal products of oxygen metabolism and are produced in excess during the course of ischemia/reperfusion through a variety of mechanism. Intracellular ROS are capable of inducing damage and, in severe cases, cell death through mitochondrial alterations leading to the release of cytochrome c (11-12), through activation of the JNK pathway (13) or by activation of nuclear factor-KB (NF-KB) transcription factors (14). The ability to control ROS is thus critical in neurodegenerative diseases, because neuronal damage occurs when the “oxidant- anti-oxidant” balances are disturbed in favor of oxidative stress (15). Generation of nitric oxide (NO), a versatile molecule in signaling processes and unspecific immune defense, is intertwined with synthesis, catabolism and transport of arginine which thus ultimately participates in the regulation of a fine-tuned balance between normal and pathophysiological consequences of NO production (16). The exact mechanisms contributing to increased production of NO in anoxia are not well established. NO induces changes in neuronal, signaling-related functions by several ways (17). NO is synthesized from arginine by nitric oxide synthase (NOS; EC 1.14.13.39), and the citrulline generated as a by-product can be recycled to arginine by successive actions of argininosuccinate synthetase (AS; EC 6.3.4.5) and argininosuccinate lyase (AL; EC 4.3.2.1) via the citrulline-NO cycle (18). Arginine in brain is also utilized by arginase (EC 3.5.3.1) for production of ornithine. Co-induction of AS, cationic amino acid transporter-2, and NOS in activated murine microglial cells (19) and co-induction of inducible NOS and arginine recycling enzymes in cytokine-stimulated PC12 cells and high output production of NO were reported (18). In our earlier study we reported the increased activities of NOS, AS and AL in kainic acid (KA) mediated excitotoxicity in rat brain (20). Thus it is hypothesized that the citulline-NO cycle enzyme activities are increased to facilitate high and continuous production of NO and increased NO may decrease the activity of GS and increase the oxidative stress in anoxia/reperfusion induced excitotoxicity. Global hypobaric hypoxia (Anoxia) is associated with many physiological and pathological conditions such as pulmonary and cardiac diseases, high altitude pathophysiology, obstructive sleep apnea, depressurization accidents and also during incidents involving anesthesia. To understand the role of citrulline-NO cycle enzymes, GS and the oxidative status in anoxia and reperfusion, NOS, AS, AL, GS and arginase activities along with the concentration of NO as nitrate /nitrite (NOx), lipid peroxidation products as Thiobarbituric acid reactive substances (TBARS) and Total antioxidant status (TAS) were estimated in cerebral cortex (CC), cerebellum (CB) and brain stem (BS) of rats subjected to anoxia (hypobaric hypoxia) and reperfusion (reoxygenation).
Materials and Methods
Male Sprague Dawley rats weighing 200 - 250 grams were used for the study. The animals had free access to food and water. Animal ethics committee and research committee of Universiti Sains Malaysia, Health campus, Kubang Kerian, Malaysia, approved the experimental design. The animals were divided into control, anoxia (global hypobaric hypoxia) and reperfusion (reoxygenation) groups (n=6 rats/group). In the Anoxia group of animals, anoxia was produced as per the procedure of Sadasivudu and Swamy (21). This method refers to global hypobaric hypoxia. The rats were placed in a desiccator whose outlet was connected to a vacuum pump and the air removed producing hypobaric conditions. About 4-5 min after the exposure of rats to hypobaric condition, the rats became lethargic and motionless. At this juncture, the rats were removed and killed by decapitation. The brains were quickly removed and the different regions (CC, CB and BS) were separated according to the procedure described by Sadasivudu and Lajtha (22). Each of the brain regions was weighed and used for the preparation of homogenates in 0.05M phosphate buffer pH 7.3. In the reperfusion group (re- oxygenated), the animals were subjected to anoxia as described for anoxia group once and after removal of animals from desiccator, they were allowed to stay at normal conditions and were given normal diet for 5 days and decapitated and the different brain regions (CC, CB and BS) were used for study. It was reported by Ananth et al (23) that 5 days showed most severe damage in a 1-21 days study after induction of excitotoxicity and hence that period (5 days) was chosen for the reperfusion group.
Enzyme assays: Total NOS activity (all isoforms of NOS: nNOS, iNOS & eNOS) was estimated by the method of Yui et al (24) as described by Swamy et al (25), in which the stable end products, NOx, were estimated using the Nitric Oxide Synthase Assay Kit from Calbiochem (Catalogue Number 482702). AS, AL activities were estimated by the modified method of Levin (26) as described by Swamy et al (25). Arginase activity was assayed according to the method of Herzfeld and Raper (27) as described by Swamy et al (24). GS activity was assayed by the method Rowe et al (28) as described by Sadasivudu et al (29).
Estimations of NO, TBARS and TAS: NO was estimated as NOx by Griess reaction after conversion of nitrate to nitrite by nitrate reductase, as described by Swamy et al (24) using the commercially available Nitric Oxide Assay Kit from Cayman Chemical Company (Catalogue number 780001; Anna Arbor, Machigan, USA). Lipid peroxidation was determined by the method of Chatterjee et al (30) by estimating TBARS. TAS was estimated according to the method of Koracevic et al (31).
Statistical analysis: Results were reported as mean ± standard deviation (SD) from 6 animals for each parameter calculated. Statistical analysis of results was done by one-way analysis of variance (ANOVA) followed by post hoc analysis using Bonferroni's test, using the SPSS software (version 12.0.1) to determine the statistical significance of difference in values between the control, anoxia and reperfusion groups. p value of < 0.05 was taken as statistically significant at 95% confidence interval.
Results
The activity of NOS (Figure 1) was increased significantly in all the three brain regions indicating increased production of NO in anoxia. In reperfusion group the activities of NOS was increased when compared to control, however it was decreased when compared to anoxia in all the brain regions tested. In anoxia group the increased activity of NOS may represent predominantly of nNOS isoform. In reperfusion group the activity may be attributed to iNOS and nNOS and increased activity may be mainly by iNOS due to expected inflammation after anoxia. The Figure 2 shows activities of AS, AL and arginase in the study. AS and AL activities increased in all the three brain regions significantly in anoxia suggesting an increased utilization of citrulline for the production of arginine in anoxia. In reperfusion group the activities of these enzymes were increased when compared to control, however they were decreased when compared to anoxia in all the brain regions tested. The activity of arginase (Figure 2) showed no significant change, indicating there was no increased utilization of arginine by this enzyme in anoxia. However in reperfusion group arginase activity was significantly increased and that may be responsible to curtail the supply of arginine for NO production in reperfusion.
Figure 1
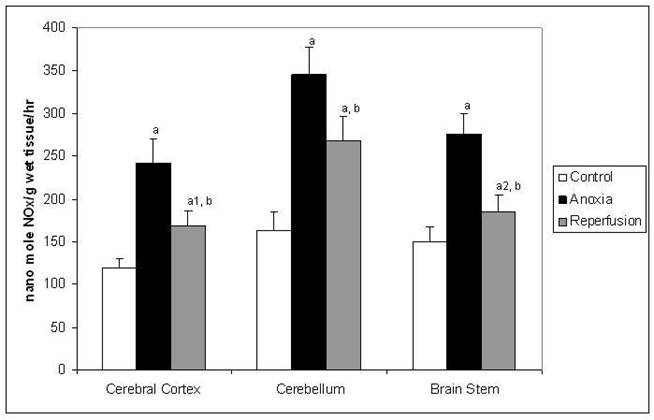
Activity of NOS in different regions of rat brain in anoxia and reperfusion. Statistical analysis was done by one-way ANOVA followed by post hoc analysis using Bonferroni's test. Values are mean ± S.D. for six animals in each group; a p < 0.001, a1 p<0.01 and a2 p<0.05 versus control group; b p <0.001 versus anoxia group.
Figure 2
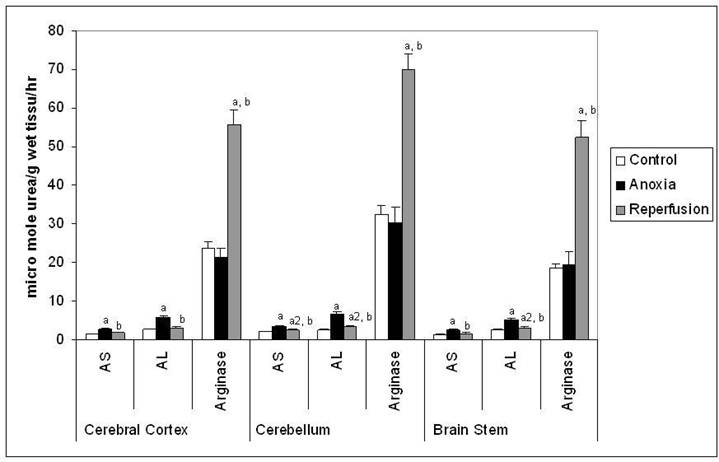
Activities of AS, AL and Arginase in different regions of rat brain in anoxia and reperfusion. Statistical analysis was done by one-way ANOVA followed by post hoc analysis using Bonferroni's test. Values are mean ± S.D. for six animals in each group; a p < 0.001, a1 p<0.01 and a2 p<0.05 versus control group; b p <0.001 versus anoxia group.
The GS activity (Figure 3) was decreased in all the tree brain regions in anoxia and showed further decrease in reperfusion group compared to control. In anoxia the possible decrease of GS may be due to the proposed modification of this enzyme by NO (32-33). In reperfusion group may be a cumulative effect of many factors such as down regulation of enzyme production and increased clearance along with the modulation by NO.
The figure 4 shows the concentration of NOx, TAS and TBARS in this study. The concentration of NOx and TBARS increased significantly in all the brain regions tested in anoxia compared to control. In reperfusion group the concentration of NOx and TBARS increased significantly when compared to control, however they were decreased when compared to anoxia in all the brain regions tested. The pattern observed for the increase in concentration of NOx in the three different brain regions was similar to that of increased NOS activity in anoxia and reperfusion groups. Concentration of TAS (Figure 4) decreased significantly in all the brain regions tested in anoxia compared to control. In reperfusion groups the decrease of TAS was lesser than that of anoxia group. The decrease in TAS and increase in TBARS levels confirms that, there is an increased oxidative stress in anoxia and reperfusion.
Figure 3
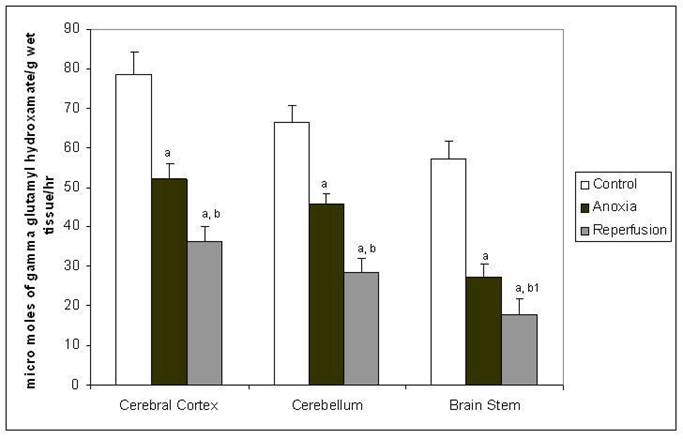
Activity of glutamine synthetase in different regions of rat brain in anoxia and reperfusion. Statistical analysis was done by one-way ANOVA followed by post hoc analysis using Bonferroni's test. Values are mean ± S.D. for six animals in each group; a p < 0.001 versus control group; b p <0.001, b1 p <0.01 versus anoxia group.
Figure 4
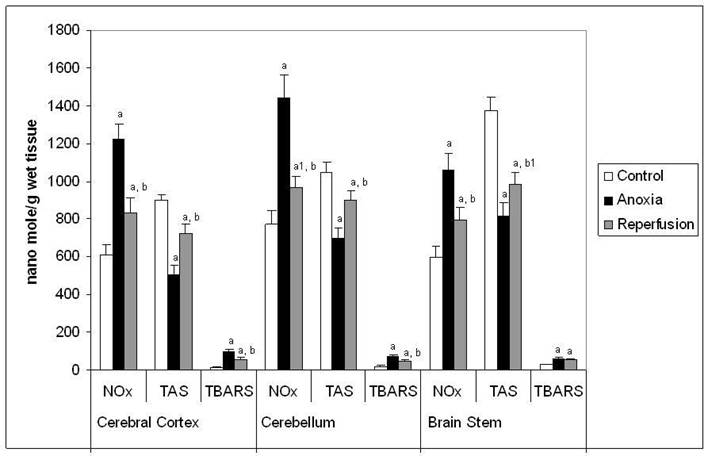
Concentration of NOxx, TASy and TBARSz in different regions of rat brain in anoxia and reperfusion. x Concentration expressed as nanomol of NOx /g wet weight of tissue. y Concentration expressed as nanomol of uric acid equivalent / g wet weight of tissue. z Concentration expressed as nanomoles of MDA equivalent / g wet weight of tissue. Statistical analysis was done by one-way ANOVA followed by post hoc analysis using Bonferroni's test. Values are mean ± S.D. for six animals in each group; a p < 0.001, a1 p<0.01 versus control group; b p <0.001, b1 p<0.01 versus anoxia group.
Discussion
Under physiological conditions, most of the glutamate in the CNS localizes to presynaptic vesicles (34) and returns there rapidly after being released during depolarizing events. However, pathological situations such as hypoxia and ischemia can lead to excessive release of glutamate and its accumulation in the extracellular space, which initiates the pathway of neuronal death known as excitotoxicity (35-36). Neuronal excitation involving the excitatory glutamate receptors is recognized as an important underlying mechanism in neurodegenerative disorders (37). In neurons, NO synthesis is stimulated by Ca2+-influx, which is induced by activation of glutamate receptors, preferentially NMDA receptor (38). The literature findings implicate neuronal NO generation in the pathogenesis of both direct and secondary excitotoxic neuronal injuries in vivo. Excitotoxicity, oxidative stress and apoptosis comprise major routs of hypoxic-ischemic neuronal death. Each route is likely activated and propagated through selective transmembrane and intracellular signaling system (39). One of the events triggered by excessive glutamate release and relevant to excitotoxicity is the production of NO (40). NO synthesis is activated cerebrovascular diseases by release of glutamate combined with inhibition of glutamate removal, which leads to NMDA receptor over activation and excess Ca2+ influx (41). The increased activity of NOS and the increased formation of NO in brain in anoxia as observed by increase in NOx concentration in this study support the earlier findings of NO involvement in pathophysiology of hypobaric hypoxia in brain (42-43). In this study the increased activity of NOS in anoxia may represent predominantly the nNOS form. In inflammatory conditions microglia and astroglia are capable of expressing iNOS (44). Following induction iNOS is known to produce large amounts of NO over prolonged periods of time, in response to many stimuli such as inflammation (45). The activity of NOS in reperfusion group may be attributed to iNOS and nNOS and the relative contribution to the increased activity in reperfusion may be mainly the iNOS form and some contribution by nNOS, as reported that nNOS expression in degenerating neurons observed through increased nNOS immunoreactivity at 5days after induction of excitotoxicity (23). Increased formation of NO by stimulated activity of NOS depends upon a continuous supply of arginine at the site of synthesis. Arginine is a semi essential amino acid and in CNS, its availability depends upon: (a) uptake from the circulating arginine and (b) recycling of citrulline to arginine by the actions of AS and AL. L-arginine is transported into synaptosomes (46), neurons (47) and astroglia (48) by the y+ CAT system. The CAT, a sodium-independent transporter, also functions as a carrier for L-ornithine in addition to L-arginine (49). Activities of AS and AL were elevated in all the brain regions of rats subjected to anoxia. The mechanism of increased activities of these enzymes is not clear. However the possibility of induction cannot be ruled out along with the modification of enzyme with unknown mechanism. Further, our observation of higher activities of NOS, AS, and AL in cerebellum suggests a higher flux of the citrulline-NO cycle in cerebellum. The functional significance of such a high flux of citrulline-NO cycle in cerebellum needs further clarification. Our results on the activities of AS and AL suggest that the citrulline-NO cycle plays a significant role in ensuring adequate supply of arginine for the increased production of NO observed in anoxia. A similar increase of NOS, AS and AL activities along with increased production of NO in KA mediated excitotoxicity was earlier reported (20).
Apart from NO synthesis L-arginine may also serve as a substrate for glutamate formation and may also provide increased substrate for arginase (50). No significant changes in the activity of arginase in the anoxic group indicate that there is no enhanced utilization of arginine by this enzyme in anoxia. However the observed increase of arginase in reperfusion group may favor decreased production of NO in this condition. Both NOS and arginase use arginine as a common substrate, and arginase may down-regulate NO production by competing with NOS for arginine (51), thus may be reducing the effects of NO in reperfusion group. The mechanism of increased activity of arginase in reperfusion is not known and may be up regulated with the supply of oxygen.
The glutamine synthetase activity is present in all parts of brain and it is equally high in cerebral cortex, cerebellum and hippocampus (33, 52). Modulation of GS activity in brain therefore important and its impairment or saturation may have pathological consequences (53). The decreased activity of GS observed in this study indicates the probable inhibition of GS by NO in anoxia. The exact mechanism of inhibition of GS by NO is not known, but it is thought to be as a covalent modification as a result of nitrosylation or nitration of tyrosin in GS (31-32, 54). It is proposed that the inhibition of GS by NO may provide prolonged availability of glutamate causing excitotoxicity in hypobaric hypoxia. Similar decrease of GS activity in KA mediated excitotoxicity was reported (20). In view of these observations the pharmacological agents who can increase GS may prove beneficial in the treatment of neurological disorders involving excitotoxicity as a result of anoxia. The decreased activity of GS in reperfusion group compared to anoxia may be a cumulative of other factors, including a down regulation of GS production and an increased clearance of this enzyme apart from modulation of GS by NO. Activity of the GS could be involved in the regulation of concentration of glutamate in the extra-cellular space of neurons with other systems, astroglial glutamate transporter-I (GLT-1), and also microglial antiporter for cystine and glutamate which may release glutamate when the demand of glutathione synthesis was increased by oxidative stress (55). From our study, the increased concentration of TBARS and decreased concentration of TAS supports the oxidative stress in anoxia.
In this study the results of citrulline - NO cycle enzymes and NOx, TAS and TBARS indicating the trend of normalization of these parameters with the supply of oxygen in reperfusion group. The increased activity of arginase in reperfusion group may be beneficial by the way of competing NOS for arginine. In a recent study using GS inhibitor, it is showed a reduced concentration of glutamine and glutamate in brain due to GS inhibition (56). The sustained decreased activity of GS in reperfusion group compared to anoxia may be a cumulative of other factors, including a down regulation of GS production and an increased clearance of this enzyme apart from modulation of GS activity by NO. The prolonged decreased GS activity may be providing benefit in reducing glutamine and glutamate concentration because the glutamine produced in the glial cells enters the neuron and converted to glutamate by glutaminase, which is described as glutamate - glutamine cycle (10, 56). Hence the decreased GS and increased arginase activities may be protective in reperfusion group.
In conclusion, this study clearly demonstrated the increased formation of NO and supports the involvement of NO in the pathophysiology of anoxia (hypobaric hypoxia) and reperfusion damage in brain. The increased activities of AS and AL indicate the effective recycling of citrulline to arginine and suggest a functional role to citrulline -NO cycle enzymes in anoxia. The decreased activity of GS in the brain regions indicate the modulation of its activity by NO and favors the prolonged availability of glutamic acid causing excitotoxicity leading to neuronal damage in anoxia. The increased concentration of TBARS and decreased concentration of TAS supports the oxidative stress in anoxia and reperfusion and suggests a possible role for anti oxidants in preventing neurodegeneration in anoxia and reperfusion injuries to brain. The increased arginase and sustained decrease of GS activity in reperfusion group are likely to be protective.
Acknowledgements
This study received support from USM- FRGS grant (A/C No: 203/PPSP/61700217). Part of the study results were presented in the 8th annual Scientific and general Meeting, College of Pathologists, Academy of Medicine, 6th - 7th June 2009, Renaissance Kota Bharu Hotel, Kelantan, Malaysia.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Coyle JT, Puttfarcken P. Oxidative stress, glutamate and neurodegenerative disorders. Science. 1993;262:689-695
2. Platt SR. The role of glutamate in central nervous system health and disease - A review. Vet J. 2007;173:278-286
3. Berliocchi L, Bano D, Nicotera P. Ca2+ signals and death programmes in neurons. Philos Trans R Soc Lond B Biol Sci. 2005;360:2255-2258
4. Back SA, Craig A, Kayton RJ, Luo NL. et al. Hypoxia-ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J Cereb Blood Flow Metab. 2007;27:334-347
5. Follett PL, Deng W, Dai W. et al. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: A protective role for topiramate. J Neurosci. 2004;24:4412-4420
6. Johnston MW. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005;15:234-240
7. Volpe JJ. Neurobiology of periventricular leukomalacia in premature infant. Pediatr Res. 2001;50:553-562
8. Sivakumr V, Ling EA, Lu J, Kaur C. Role of glutamate and its receptors and insulin-like growth factors in hypoxia induced periventricular white matter injury. Glia. 2010;58:507- 523
9. Szatkowski M, Attwell D. Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci. 1994;17:359-365
10. Van der berg CJ, Garfinkel D. A stimulation study of brain compartments, metabolism of glutamate and related substances in mouse brain. Biochem J. 1971;123:211-218
11. Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria, implications for Parkinson's disease. J Neurochem. 1999;73:1127-1137
12. Halestrap AP, Doran E, Gillespie JP, O'Toolee A. Mitochondria and cell death. Biochem Soc Trans. 2000;28:170- 177
13. Tournier C, Hes P, Yang DD. et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870-874
14. Luo Y, Hattori A, Munoz J. et al. Intrastriatal dopamine injection induces apoptosis through oxidation-involved activation of transcription factors AP-1 and NK-kappaB in rats. Mol Pharmacol. 1999;56:254-264
15. Maalouf M, Sullivan PG, Davis L. et al. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007;145:256-264
16. Wiesinger H. Arginine metabolism and the synthesis of nitric oxide in the nervous system. Prog Neurobiol. 2001;64:365-391
17. Prast H, Philippu A. Nitric oxide as modulator of neuronal function. Prog Neurobiol. 2001;64:51-68
18. Zhang WY, Gotoh T, Oyadomari S, Mori M. Coinduction of inducible nitric oxide synthase and arginine recycling enzymes in cytokine-stimulated PC 12 cells and high output production of nitric oxide. Mol Brain Res. 2000;83:1-8
19. Kawahara K, Gotoh T, Oyadomari S. et al. Co-induction of argininosuccinate synthetase, cationic amino acid transporter-2, and nitric oxide synthase in activated murine microglial cells. Mol Brain Res. 2001;90:165-173
20. Swamy M, Sirajudeen KNS, Chandran G. Nitric oxide [NO] citrulline-NO cycle enzymes, glutamins synthetase and oxidative status in kainic acid-mediated excitotoxicity in rat brain. Drug Chem Toxicol. 2009;32:326-331
21. Sadasivudu B, Swamy M. Possible occurrence of ornithine-ω- aminotransferase in GABAergic neurons. Neurochem Res. 1984;9:1593-1598
22. Sadasivudu B, Lajtha A. Metabolism of amino acids in incubated slices of mouse brain. J. Neurochem. 1970;17:1299-1311
23. Ananth C, Thameem Deen S, Gopalakrishnakone P, Kaur C. Domoic acid-induced neuronal damage in the rat hippocampus: Changes in apoptosis related genes (Bcl-2, Bax, Caspase-3) and microglial response. J Neurosci Res. 2001;66:177- 190
24. Yui Y, Hattori R, Kosuga K. et al. Calmodulin-independent nitric oxide synthase from rat polymorphonuclear neutrophils. J Biol Chem. 1991;266:3369-3371
25. Swamy M, Adlin ZZ, Chandran G. et al. Effect of acute ammonia toxicity on nitric oxide (NO), citrulline-NO cycle enzymes, arginase and related metabolites in different regions of rat brain. Neurosci Res. 2005;53:116-122
26. Levin B. Hereditary metabolic disorders of urea cycle. In: (ed.) Bodensky O, Latner AL. Advances in Clinical Chemistry, Vol 14. New York: Academic Press. 1971:66
27. Herzfeld A, Raper SM. The heterogeneity of arginase in rat tissues. Biochem J. 1976;153:469-478
28. Rowe WB, Ronzio RA, Wellner VP, Meister A. Glutamine synthetase (Sheep brain). In: (ed.) Tabor H, Tabor CW. Methods in Enzymol, Vol XVII Part A. New York: Academic Press. 1970:900-910
29. Sadasivudu B, Zahida Nasreen, Swamy M. Functional significance of the activities of gutaminase and ornithine-ω- aminotransferase in rat brain. Neurochem Int. 1985;7:449-454
30. Chatterjee PK, Cuzzocrea S, Brown PA. et al. Tempol, a membrane-permeable radical scavenger, reduces oxidant stress-mediated renal dysfunction and injury in the rat. Kidney Int. 2000;58:658-673
31. Koracevic D, Koracevic G, Djordjevic V. et al. Method for the measurement of antioxidant activity in human fluids. J Clin Pathol. 2000;54:356-361
32. Kosenko E, Llansola M, Montoliu C. et al. Glutamine synthetase activity and glutamine content in brain: modulation by NMDA receptors and nitric oxide. Neurochem Int. 2003;43:493-499
33. Rose C, Felipo V. Limited capacity for ammonia removal by brain in chronic liver failure: potential role of nitric oxide. Metab Brain Dis. 2005;20:275-283
34. Naito S, Ueda T. Adenosine triphosphate-dependent uptake of glutamate into protein I-associated synaptic vesicles. J Biol Chem. 1983;258:696-699
35. Dienel G, Hertz L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia. 2005;50:362-388
36. Rothman SM, Olney JW. Glutamate and the pathophysiologyof hypoxic-ischemic brain damage. Ann. Neurol. 1986;19:105-111
37. Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenisis of neurodegenerative diseases. Acta Pharmocol. Sin. 2009;30:379-387
38. Radenovic L, Selakovic V. Differential effects of NMDA and AMPA/Kainate receptor antagonists on nitric oxide production in rat brain following intrahippocampal injection. Brain Res Bull. 2005;67:133-141
39. Won SJ, Kim DY, Gwang BJ. Cellular and molecular pathways of ischemic neuronal death. J. Biochem. Mol. Biol. 2002;35:67-86
40. Parathath SR, Gravanis I, Tsirka SE. Nitric Oxide Synthase Isoforms Undertake Unique Roles During excitotoxiciti. Stroke. 2007;38:1938-1945
41. Law A, Gauthier S, Quirion R. Say NO to Alzheimer's disease, the putative links between nitric oxide and dementia of Alzheimer's type. Brain Res Rev. 2001;35:73-96
42. Serrano J, Encinas JM, Salas E. et al. Hypobaric hypoxia modifies constitutive nitric oxide synthase activity and protein nitration in the rat cerebellum. Brain Res. 2003;976:109-119
43. Serrano J, Encinas JM, Salas E. et al. Effect of hypobaric hypoxia on the nitric oxide system of the rat cerebral cortex: Protective role of nitric oxide inhibitors. Neuroscience. 2006;142:799-808
44. Nomura EA, Kitamura Y. Inducible nitric oxide synthase in glial cells. Neurosci Res. 1993;18:103-107
45. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1974:323-350
46. Aldridge CR, Collard KJ. The characteristics of arginine transport by rat cerebellar and cortical synaptosomes. Neurochem Res. 1996;21:1539-1546
47. Westergaard N, Beart PM, Schousboe A. Transport of L-[3H] arginine in cultured neurons: characteristics and inhibition by nitric oxide synthase inhibitors. J Neurochem. 1993;61:364-367
48. Schmidlin A, Wiesinger H. Transport of L-arginine in cultured glial cells. Glia. 1994;11:262-268
49. Hosokawa H, Sawamura T, Kobayashi S. et al. Cloning and characterization of a brain-specific amino acid transporter. J Biol Chem. 1997;272:8717-8722
50. Bommarius AS, Makryaleas K, Drauz K. An enzymatic route to L-ornithine from L-arginine--activation and stabilization studies on L-arginase. Biomedica Biochimica Acta. 1991;50:S249-S255
51. Mori M. Regulation of Nitric oxide synthesis and apoptosis by arginase and arginine recycling. J Nutr. 2007;137:1616S-1620S
52. Girard G, Giguere J-F, Butterworth RF. Region selective reductions in activities of glutamine synthetase in rat brain following portacaval anastomosis. Metab Brain Dis. 1993;8:207-215
53. Rodrigo R, Felipo V. Control of brain glutamine synthesis by NMDA receptors. Front Biosci. 2007;12:883-890
54. Bidmon HJ, Gorg B, Palomero-Gallagher N, Schleicher A. Glutamine synthetase becomes nitrated and its activity is reduced during repetitive seizure activity in the pentylenetetrazole model of epilepsy. Epilepsia. 2008;49:1733-1748
55. Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205-1213
56. Ghoddoussi F, Galloway MP, Jambekar A. et al. Methionine sulfoximine, an inhibitor of glutamine synthetase, lowers brain glutamine and glutamate in a mouse model of ALS. J Neurol Sci. 2010;290:41-47
Author contact
![]() Corresponding author: Dr. Mummedy Swamy, Department of Chemical Pathology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia. E-mail: mswamyusm.my, mummedysco.in. Fax: +609-765 3370
Corresponding author: Dr. Mummedy Swamy, Department of Chemical Pathology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia. E-mail: mswamyusm.my, mummedysco.in. Fax: +609-765 3370
Citation styles
APA
Swamy, M., Salleh, M.J.M., Sirajudeen, K.N.S., Yusof, W.R.W., Chandran, G. (2010). NITRIC OXIDE (NO), CITRULLINE - NO CYCLE ENZYMES, GLUTAMINE SYNTHETASE AND OXIDATIVE STRESS IN ANOXIA (HYPOBARIC HYPOXIA) AND REPERFUSION IN RAT BRAIN. International Journal of Medical Sciences, 7(3), 147-154. https://doi.org/10.7150/ijms.7.147.
ACS
Swamy, M.; Salleh, M.J.M.; Sirajudeen, K.N.S.; Yusof, W.R.W.; Chandran, G. NITRIC OXIDE (NO), CITRULLINE - NO CYCLE ENZYMES, GLUTAMINE SYNTHETASE AND OXIDATIVE STRESS IN ANOXIA (HYPOBARIC HYPOXIA) AND REPERFUSION IN RAT BRAIN. Int. J. Med. Sci. 2010, 7 (3), 147-154. DOI: 10.7150/ijms.7.147.
NLM
Swamy M, Salleh MJM, Sirajudeen KNS, Yusof WRW, Chandran G. NITRIC OXIDE (NO), CITRULLINE - NO CYCLE ENZYMES, GLUTAMINE SYNTHETASE AND OXIDATIVE STRESS IN ANOXIA (HYPOBARIC HYPOXIA) AND REPERFUSION IN RAT BRAIN. Int J Med Sci 2010; 7(3):147-154. doi:10.7150/ijms.7.147. https://www.medsci.org/v07p0147.htm
CSE
Swamy M, Salleh MJM, Sirajudeen KNS, Yusof WRW, Chandran G. 2010. NITRIC OXIDE (NO), CITRULLINE - NO CYCLE ENZYMES, GLUTAMINE SYNTHETASE AND OXIDATIVE STRESS IN ANOXIA (HYPOBARIC HYPOXIA) AND REPERFUSION IN RAT BRAIN. Int J Med Sci. 7(3):147-154.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.