Impact Factor ISSN: 1449-1907
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Ligation methods
Arguments for the...
Synthesis of diene compounds
Synthesis of tetrazine diene...
Functionalization of tetrazines...
Synthesis of dienophile compounds
Synthesis of the dienophile by...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2010; 7(1):19-28. doi:10.7150/ijms.7.19 This issue Cite
Research Paper
The Diels-Alder-Reaction with inverse-Electron-Demand, a very efficient versatile Click-Reaction Concept for proper Ligation of variable molecular Partners
Manfred Wiessler1, Waldemar Waldeck2, Christian Kliem1, Ruediger Pipkorn3, Klaus Braun1 ![]()
1. German Cancer Research Center, Dept. of Imaging and Radiooncology, INF 280, D-69120 Heidelberg, Germany
2. German Cancer Research Center, Division of Biophysics of Macromolecules, INF 580, D-69120 Heidelberg, Germany
3. German Cancer Research Center, Central Peptide Synthesis Unit, INF 580, D-69120 Heidelberg, Germany
Received 2009-1-27; Accepted 2009-11-25; Published 2009-12-5
Abstract
The ligation of active pharmaceutical ingredients (API) for working with image processing systems in diagnostics (MRT) attracts increasing notice and scientific interest. The Diels-Alder ligation Reaction with inverse electron demand (DARinv) turns out to be an appropriate candidate. The DARinv is characterized by a specific distribution of electrons of the diene and the corresponding dienophile counterpart. Whereas the reactants in the classical Diels-Alder Reaction feature electron-rich diene and electron-poor dienophile compounds, the DARinv exhibits exactly the opposite distribution of electrons. Substituents with pushing electrones increase and, with pulling electrons reduce the electron density of the dienes as used in the DARinv.
We report here that the DARinv is an efficient route for coupling of multifunctional molecules like active peptides, re-formulated drugs or small molecules like the alkyalting agent temozolomide (TMZ). This is an example of our contribution to the "Click chemistry" technology. In this case TMZ is ligated by DARinv as a cargo to transporter molecules facilitating the passage across the cell membranes into cells and subsequently into subcellular components like the cell nucleus by using address molecules. With such constructs we achieved high local concentrations at the desired target site of pharmacological action. The DARinv ligation was carried out using the combination of several technologies, namely: the organic chemistry and the solid phase peptide synthesis which can produce 'tailored' solutions for questions not solely restricted to the medical diagnostics or therapy, but also result in functionalizations of various surfaces qualified amongst others also for array development.
We like to acquaint you with the DARinv and we like to exemplify that all ligation products were generated after a rapid and complete reaction in organic solutions at room temperature, in high purity, but also, hurdles and difficulties on the way to the TMZ-BioShuttle conjugate should be mentioned.
With this report we would like to stimulate scientists working with the focus on "Click chemistry" to intensify research with this expanding DARinv able to open the door for new solutions inconceivable so far.
Keywords: Click-Chemistry, Cycloaddition, Ligation chemistry, Linker Systems, Adaptor Systems, inverse Diels Alder Reaction, Tetrazines, Therapy, Triazines, Diagnostics
Introduction
Due to small molecular weight traditional chemotherapeutics reach the DNA target by diffusion. But the lack of specificity necessitates highly application doses and results in adverse reactions which hampers the therapeutic outcome. Modern high specific drug formulations, consisting of DNA and derivatives, could circumvent these insurmountable obstacles; unfortunately their physico-chemical properties as well as in the big molecular weight result in a barely noticeable diffusibility, insufficient for therapeutic local concentrations or for an intravital diagnostics imaging. Both approaches need a proper coupling of drugs or intravital contrast agents to carrier molecules for the passage across cellular membranes and subsequently for the transport into subcellular components like the cell nucleus for imaging of molecular processes at the transcriptional level. Therefore rapid and selective ligation of pharmacologically active molecules or modern diagnostics increasingly comes to the fore of the scientific research and poses a great challenge to chemists.[1] The cornucopia of chemistry harbours a collection of chemical reactions whose mechanisms were identified, characterized and collected during decades, offering promising solutions.
Ligation methods
A series of qualified reactions were compiled by Sharpless in its concept for the Click-Chemistry [2] dealing with consistently and rapidly running chemical ligation reactions without side reactions and with inexpensive manufacturing of the educts. As Sharpless wrote, the electrocyclic reactions rank amongst prime examples of the Click-Chemistry, since they dispose the sufficient driving force and also the required selectivity. This 1,3-dipolar cycloaddition, well investigated during the last years, is a representative reaction mechanism, based on Huisgen's work. [3] Nowadays the ligation reaction modified by Sharpless is considered as "Cream of the Crop" [4] setting the benchmark compared to all other competing reactions. However the 1,3-dipolar cycloaddition needs long reaction times and additionally stringent conditions like high temperatures. Using the catalytically accelerated reaction variant introduced by Sharpless, only few hours at room temperature are required [5] as shown with a plenty of reaction examples. [6-14] A second well characterized reaction is the Staudinger Ligation, mainly developed by Bertozzi [15, 16] based on the well documented reaction of phosphines with azides resulting in phosphoamide formation and nitrogen elimination. In contrast to this, in the intramolecular variant, the phosphine group is eliminated and a proper amidation is established. The third reaction useful for the Click-Chemistry is the thioester-method. [17-21]
Several examples underlining the relevance of the classical DAR for ligation of molecules are documented. [22-25]
It is quite remarkable that the DAR fulfills all criteria of the Click-Chemistry, but generally the reaction rate is very low at room temperature. The main drawback of the DAR is not only due to its reversibility but due to the extent of equilibrium formation between diene and dienophile compounds for one or the other Diels-Alder-product. In principal formations of exo- and endo-products are possible, at which the endo-product is thermodynamically favoured. A further restriction of the DAR may be expected under physiological conditions using furans as dienes and maleinimides as dienophiles for example and thus it is difficult to carry out the DAR e.g. in the presence of proteins with unrestricted SH-groups.
As specified above, this clubfoot combines several methods: it needs a catalytic support and stringent conditions. Purification and removing the catalysts turned out to be exceedingly difficult and the present reverse reactions to the corresponding educts exacerbate the application in biological systems.
Arguments for the DARinv as ligation method
Circumventing the DAR's drawbacks as mentioned above, the irreversible inverse Diels-Alder-Reaction process (DARinv) has been predicted and was already documented in 1959. [26]
Since 1960 the DARinv was investigated intensively, particularly by Sauer, Neunhoeffer and Seitz in 2001 [27-32] Boger and Snyder succeeded in the synthesis of nature identical materials using this technology. [33, 34] The DARinv features a specific distribution of the electron density in its reacting agents: Whereas the reactants in the classical Diels-Alder Reaction possess electron-rich dienes and electron-poor dienophile compounds, the DARinv exhibits exactly the opposite distribution of electrons.
This reaction (schema/table 1) fulfils all the criteria and lives up to its name “Click”-Reaction. [26, 35, 36] The careful choice of the reactants is the linchpin of the DARinv.
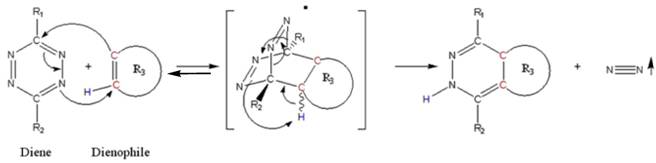
(Scheme 1) The Diels-Alder-Reaction with inverse electron demand (DARinv) is shown. R1 and R2 represent different functional moieties harbouring -I and/or -M effects on the diene A., which induce a decrease of the electron density of the tetrazine ring. Whereas in contrast the R3 features a +I effect resulting in a relative high electron densitiy in the dienophile compound. The stepwise reaction from A and B results in the stereoisomers C and D, which can be attributed to the two different variants of intermediates (bracketed) after elimination of molecular nitrogen. As shown here the reverse reaction is impossible.
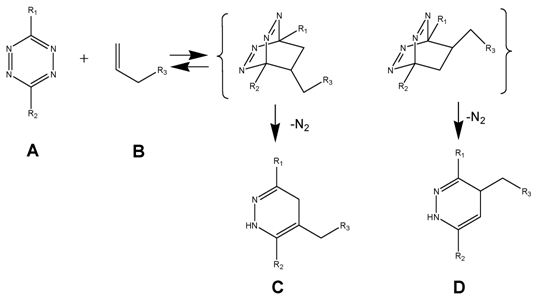
In the following we report the synthesis of functionalized dienes and dienophiles, for use as reactant for the ligation of pharmacological active substances like temozolomide (TMZ) by the DARinv recently documented in our TMZ-reformulation's studies. [37-39]
Using tetrazine derivative A (diene), the primary adduct of the DARinv, stabilizes C and D by eliminating nitrogen under formation of colorless dihydropyridazine derivatives and a reverse reaction is excluded [13] in contrast to the classical DAR. During the reaction initially the dissociation of the nitrogen from the primary adduct leads to the 4,5-dihydropyridazine product.
As generally known, the impetus of the Staudinger-Ligation originates from the dissociation of the nitrogen from the primary adduct of phosphine and azide. Insofar a formal similarity exists between both the Staudinger-Ligation and the Diels-Alder-Reaction with inverse electron demand (as elucidated in scheme/table 1). As shown, the double bonds of the stable dihydro-pyridazines exist in a crossed conjugation, an oxidation reaction to the corresponding pyridazines barely occurs in exceptional cases without addition of oxidants. In order to obtain a homogeneous reaction-product the dehydrogenation can be performed by chloranil.
Synthesis of diene compounds
The most frequently used diene in the DARinv is the easily available 1,2,4,5-tetrazine-3,6-dicarbonic acid-dimethylester 5, which can be produced in three-steps starting with diazo etylacetate 1. The esters 2 and 4 were already synthesized first by Curtius in Heidelberg. [40] The diazo ester and the hydrazine molecule 4 were isolated and described by Sauer [41] (as illustrated in scheme/table 2). Since with DARinv almost all molecules of this diester undergo a quantitative reaction within minutes and at room temperature, this method was proposed by Nenitzescu for estimation of terminal double bonds using volumetric methods with 5. [42] The broad range of measured reaction rates is a characteristic feature of molecule 1,2,4,5-tetrazine-3,6-dicarbonic acid-dimethylester 5.
(Scheme 2) Synthesis of the tetrazine dicarbonic acid 5. Reagents and conditions: are described in the methods section The reaction steps were initiated and carried out in i) 1, a) 50% NaOH, b) H2SO4; ii) NaNO2 in glacial acetic acid; iii) SOCl2, MeOH; NaNO2 iv) R-NH2; NaNO2, glacial acetic acid.
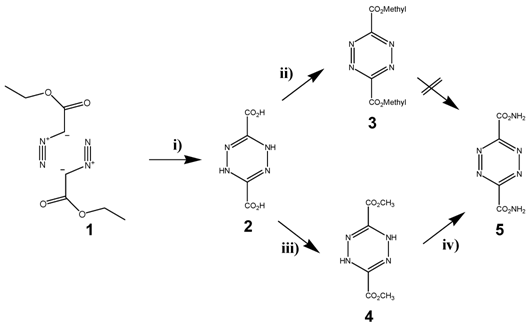
Synthesis of tetrazine diene compounds
It became evident that especially modified esters or acidic amides should be considered as appropriate tetrazine derivatives because of the reactivity of two carbonyl groups of the tetrazine derivative molecule 5 and the electro negativity is crucial for maintaining the high diene-activity. The stability of functionalized tetrazine esters, however, proved to be insufficient, whereas functionalized tetrazine amides provide the ability for the synthesis of many functionalized derivatives. They can be obtained via the dihydrotetrazine-amides followed by oxidation. Nevertheless the use of these tetrazine derivatives as described in schema/table 2 turned out to be problematic for two reasons: 1) their insufficient stability and 2) the poor solubility in aqueous solution obviate applications in living systems. The chemical reaction of the tetrazine 5 in water or with amines as nucleophiles did not yield a nucleophilic substitution, but exclusively an unexpected a ring opening at the ester groups was observed. [43] This sensitivity against nucleophiles seems to be also the cause of the low stability of these tetrazine derivatives in aqueous solution. Because of these chemical decomposition processes, tetrazines modified with the dicarboxylic acid 3 are not qualified for ligation under conditions of the solid phase peptide synthesis (SPPS) as well as under physiological conditions as proven in our experiments.
Considerations to circumvent these limitations the development gives rise to the synthesis of tetrazines aryl substituted featuring -I attributes. The colour change during DARinv of a tetrazine (magenta) to diazine (yellow) under degassing nitrogen occurs rapidly. [44]. The synthesized diaryl-tetrazines are summarized in scheme/table 2.
Functionalization of tetrazines with temozolomide -TMZ
Indeed the situation of the synthesis of a further group of dienes used in our TMZ-BioShuttle studies is unequally catchier and the synthesis of functionalized tetrazines highly suitable for the DARinv poses an experimental challenge as clearly and accessibly described in [37].
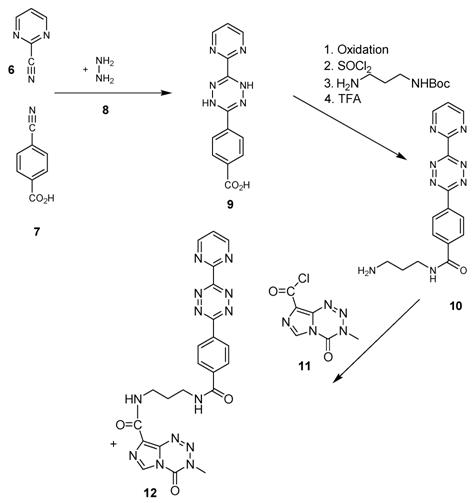
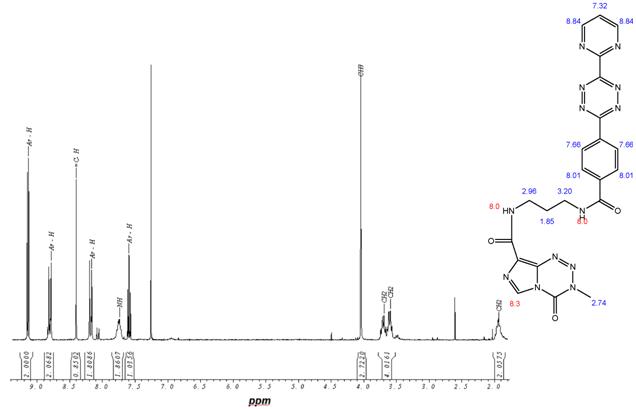
This open question for the synthesis of tetrazine-based dienes in aqueous solution is answered below. The possibility of the synthesis of tetrazines functionalized with aryl compounds is attractive and could be successful. As illustrated in schema/table 3 the synthesis worked via the following educts: 2-cyanopyrimidine 6 and 4-cyanobenzoic acid 7 react with 80% aqueous hydrazine 8 to the intermediate 3,6-diaryl-1,2-dihydro-1,2,4,5-tetrazine 9 in 40 to 50 % yield. The oxidation to the corresponding 1, 4-diaryl-1,2,4,5-tetrazine is next reaction step followed by the conversion to the acid chloride with thionyl dichloride which in turn was reacted with the Boc-mono-protected 1,3-propylenediamine to the propyleneamine substituted acid amide 10. After deprotection with TFA the amino group was transferred with the acid chloride derivative of the TMZ 11 to the TMZ-diaryl-tetrazine 12 a diene compound poised for the DARinv. The corresponding NMR H spectra are shown in the figures 2-5.
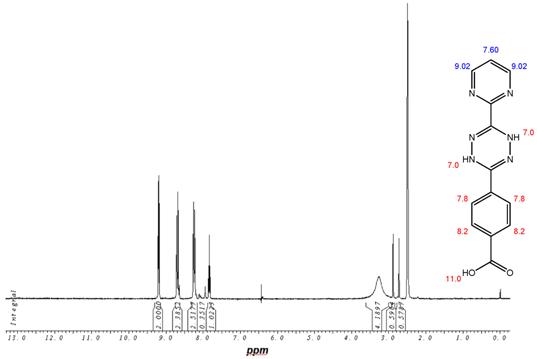
(Schema 3) Synthesis of the Temozolomide derivative 12 capable for the ligation via DARinv: The 1,3 diaminopropyl modified 4-diaryl-3,8-dihydro-1,2,4,5-tetrazine 10 is reacted with the acid chloride derivative of the TMZ.
1H-NMR-Spectrum of the 9 in D6-DMSO. The structure illustrates the shift calculation for protons of the compound with ChemDraw Ultra 2004. (Numbers indicate the predicted shift of the signals in ppm; quality of estimation is indicated in colour: blue = good, red = rough)
1H-NMR-Spectrum of the oxidised 9 in D6-DMSO. The structure illustrates the shift calculation for protons of the compound with ChemDraw Ultra 2004. (Numbers indicate the predicted shift of the signals in ppm; quality of estimation is indicated in colour: blue = good, red = rough). Insert indicates the detailed peak analysis for the signals found together with the coupling constants measured.
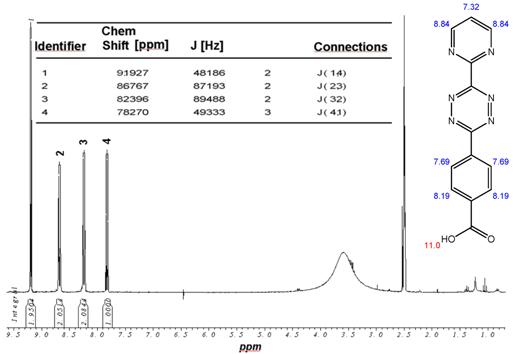
1H-NMR-Spectrum of the ammonium salt of 9 in D6-DMSO. The structure illustrates the shift calculation for protons of the compound with ChemDraw Ultra 2004. (Numbers indicate the predicted shift of the signals in ppm; quality of estimation is indicated in colour: blue = good, red = rough).
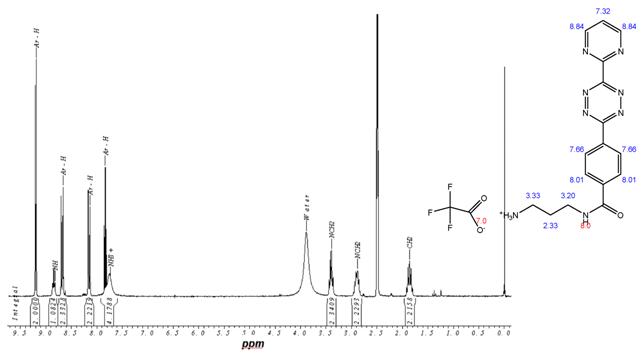
1H-NMR-Spectrum of 9 in CDCl3. The structure illustrates the shift calculation for protons of the compound with ChemDraw Ultra 2004. (Numbers indicate the predicted shift of the signals in ppm; quality of estimation is indicated in colour: blue = good, red = rough)
Synthesis of dienophile compounds
It should be mentioned that a lot of educts harbouring terminal double bonds or bonds in ring systems are commercially available or are easily to prepare. By this means a wide range of dienophilic compounds is available for DARinv.
In order to obtain reaction times in the range of minutes for the DARinv, the reactivity of the dienophile is decisive for the rapid reaction process besides the reactivity of the tetrazines as reactants. The allyl-group, a component of numerous chemical compounds which are easily accessible indeed may offer dienophile-activity, but nevertheless it is not qualified for a rapid chemical reaction. Therefore symmetric dienophiles with higher reactivity should be favoured.
In 1966 Sauer documented a very high dienophile activity of double bonds in cyclic ring systems adjoining to the terminal double bonds. [45] Incipient with strained rings like cyclopropene which posesses a very high dienophile reactivity, the reaction rate is reciprocally proportional to the ring's size; the minimum is reached with the six-atom ring, and in larger rings the reaction rate is slightly increasing. Derivatives of the cyclobutene dispose still a sufficient stability as well as excellent dienophile reactivity.
Synthesis of the dienophile by the Reppe process
A tetracyclic anhydride, synthesized and well described by Reppe (best-known under the name of Reppe-Anhydride) turned out to be the ideal compound for our purpose 15 (Figure 1). It is easily available by the classic Diels-Alder-Reaction of cyclooctatetraene (COT) 13 and maleic anhydride 14. [46-48] Additionally to the anhydride function this tetracyclic compound possesses a reactive double bond inside of the cyclobutene ring. The presence of a further double bond inside of the cyclohexene ring is not of relevance due to the inactivity of this double bond in the DARinv. The anhydride ring of the tetracyclic compound can be converted to the substituted corresponding imides, whereas the yield is nearly quantitative and the product can be purified easily by recrystallization (scheme/table 4). Further ring systems, dedicated for chemical reactions as described above, are the cyclobutene-3,4-dicarboxylic acid anhydride [49, 50], as well as the commercially available exo- and endo-norbornen-anhydrides.
Simplified mechanistic illustration of the electron rearrangement during DARinv. While the first step of association between diene and dienophile is reversible, the release of nitrogen during rearrangement of the intermediate product is irreversible and switches the reaction to the product side.
(Scheme 4) Synthesis of a versatile building block for modification of peptides. The syntheses of the Reppe-Anhydride 15 and the corresponding Boc-Lys derivative 16 are described [38].
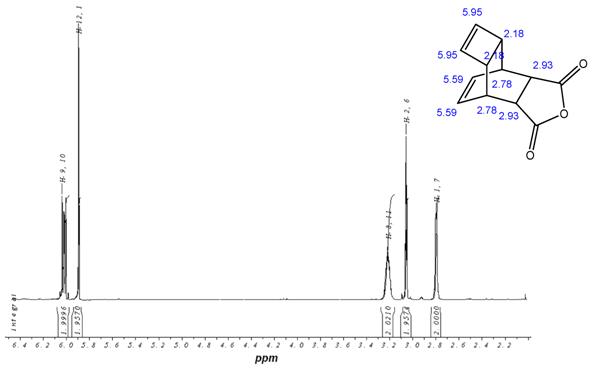
1H-NMR-Spectrum of the Reppe Anhydride 15 in CDCl3. The structure illustrates the shift calculation for protons of the compound with ChemDraw Ultra 2004. (Numbers indicate the predicted shift of the signals in ppm; quality of estimation is indicated in colour: blue = good, red = rough)
Conclusion
The DARinv chemistry can extend the ligation methods of functional molecules or genetic materials as a cargo to carrier and address-molecules to realize high local concentrations of active substances and is able to circumvent the barriers on the way to a save and efficient transfer of therapeutic and / or diagnostics cargos into target cells and tissues. It is important to point out that this DARinv technology is not restricted to application and easier handling of the BioShuttle delivery platform and of the other related carrier molecules. An enhancement towards functionalization of surfaces and polymers by a proper ligation at surfaces like arrays is so realizable. Depending on the scientific subject and formulation of the scientific project, the coupling of the diene as well as the dienophile at a surface could be demonstrated.
The following points support this technology:
- The presented kinetic data with high reaction rates demonstrate the potential of the Diels-Alder-Reaction with inverse electron demand (DARinv) as method of choice for ligation of molecules.
- The reaction process could be easily monitored by use of photometrical methods with a decreasing absorption maximum at 520 nm, which is typical for tetrazines.
- We could demonstrate that the DARinv features all the conditions for the successful “Click”-Chemistry and as a consequence turns out to be a dedicated tool for ligation reactions not restricted to the medical and pharmaceutical science.
The advantage of DARinv lies in the
- compounds's accessibility [39],
- the high and quantitative reaction rate,
- the potential for selective multiple reactions at the identical molecule,
- the easy monitoring of the chemical reaction and
- the feasibility of the reaction at surfaces.
With this report we like to emphasize the great potential of the DARinv technology exemplarily documented in the field of drug-re-formulation which dramatically increases the therapeutic potential of classic drugs as exemplarily pointed with the alkylating agent temozolomide (TMZ). It also enhanced the therapeutic spectrum in malignant gliomas or in hormone-refractory prostate cancer [37-39]. Additionally this DARinv technology attracts increasing notice to further medical applications, especially in oncological diagnostics and therapy at the molecular level.
Acknowledgements
This work was supported in part by grant from the Deutsche Krebshilfe Foundation (Project No. 106335).
We cordially thank Gabriele Mueller, Peter Lorenz, and Heinz Fleischhacker for their dedication and their technical assistance in the development of this concept.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Kohn M, Breinbauer R. The Staudinger ligation-a gift to chemical biology. Angew Chem Int Ed Engl. 2004;43:3106-16
2. Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed Engl. 2001;40:2004-21
3. Huisgen R. Theory of 1,3-Dipolar Cycloadditions. In: (ed.) Padwa A. 1,3-Dipolar Cycloaddition Chemistry. New York: Wiley. 1984:1-176
4. Rostovtsev VV, Green LG, Fokin VV. et al. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596-9
5. Chan TR, Hilgraf R, Sharpless KB. et al. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Organic Letters. 2004;6:2853-5
6. Bock VD, Hiemstra H, van Maarseveen JH. Cu-I-catalyzed alkyne-azide "click" cycloadditions from a mechanistic and synthetic perspective. European Journal of Organic Chemistry. 2005:51-68
7. Tornoe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. Journal of Organic Chemistry. 2002;67:3057-64
8. Rozkiewicz DI, Janczewski D, Verboom W. et al. "Click" chemistry by microcontact printing. Angew Chem Int Ed Engl. 2006;45:5292-6
9. Parrish B, Breitenkamp RB, Emrick T. PEG- and peptide-grafted aliphatic polyesters by click chemistry. J Am Chem Soc. 2005;127:7404-10
10. Diaz DD, Punna S, Holzer P. et al. Click chemistry in materials synthesis. 1. Adhesive polymers from copper-catalyzed azide-alkyne cycloaddition. Journal of Polymer Science Part A-Polymer Chemistry. 2004;42:4392-403
11. Whiting M, Muldoon J, Lin YC. et al. Inhibitors of HIV-1 protease by using in situ click chemistry. Angewandte Chemie-International Edition. 2006;45:1435-9
12. Chi YS, Lee JK, Lee K. et al. Biosurface Organic Chemistry: Interfacial Chemical Reactions for Applications to Nanobiotechnology and Biomedical Sciences. Bull Korean Chem Soc. 2005;26:361-9
13. Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discov Today. 2003;8:1128-37
14. Kohn M, Wacker R, Peters C. et al. Staudinger ligation: a new immobilization strategy for the preparation of small-molecule arrays. Angew Chem Int Ed Engl. 2003;42:5830-4
15. Chandra RA, Douglas ES, Mathies RA. et al. Programmable cell adhesion encoded by DNA hybridization. Angew Chem Int Ed Engl. 2006;45:896-901
16. Soellner MB, Nilsson BL, Raines RT. Staudinger ligation of alpha-azido acids retains stereochemistry. Journal of Organic Chemistry. 2002;67:4993-6
17. Camarero JA, Kwon Y, Coleman MA. Chemoselective attachment of biologically active proteins to surfaces by expressed protein ligation and its application for "protein chip" fabrication. J Am Chem Soc. 2004;126:14730-1
18. Bang D, Pentelute BL, Kent SBH. Kinetically controlled ligation for the convergent chemical synthesis of proteins. Angewandte Chemie-International Edition. 2006;45:3985-8
19. Yeo DSY, Srinivasan R, Chen GYJ. et al. Expanded utility of the native chemical ligation reaction. Chemistry-A European Journal. 2004;10:4664-72
20. Hill KW, Taunton-Rigby J, Carter JD. et al. Diels-Alder bioconjugation of diene-modified oligonucleotides. Journal of Organic Chemistry. 2001;66:5352-8
21. Seelig B, Jaschke A. Site-specific modification of enzymatically synthesized RNA: Transcription initiation and Diels-Alder reaction. Tetrahedron Letters. 1997;38:7729-32
22. Gawalt ES, Mrksich M. A substituent effects study reveals the kinetic pathway for an interfacial reaction. Journal of the American Chemical Society. 2004;126:15613-7
23. Pozsgay V, Vieira NE, Yergey A. A method for bioconjugation of carbohydrates using Diels-Alder cycloaddition. Organic Letters. 2002;4:3191-4
24. Tona R, Haner R. Synthesis and bioconjugation of diene-modified oligonucleotides. BIOCONJUGATE CHEMISTRY. 2005;16:837-42
25. Graham D, Grondin A, McHugh C. et al. Internal labeling of oligonucleotide probes by Diels-Alder cycloaddition. Tetrahedron Letters. 2002;43:4785-8
26. Carboni RA, Lindsey RV. Reactions of Tetrazines with Unsaturated Compounds - A New Synthesis of Pyridazines. J Americ Chem So. 1959;81:4342-6
27. Panek JS, Zhu B. Synthesis of aromatic 1,2-diazines by inverse electron demand Diels-Alder reaction of polymer-supported 1,2,4,5-tetrazines. Tetrahedron Letters. 1996;37:8151-4
28. Sauer J, Bauerlein P, Ebenbeck W. et al. [4+2] cycloadditions of 1,2,4,5-tetrazines and cyclopropenes - Synthesis of 3,4-diazanorcaradienes and tetracyclic aliphatic azo compounds. European Journal of Organic Chemistry. 2001:2629-38
29. Sauer J, Pabst GR, Holland U. et al. 3,6-bis(2H-tetrazol-5-yl)-1,2,4,5-tetrazine: A versatile bifunctional building block for the synthesis of linear oligoheterocycles. European Journal of Organic Chemistry. 2001:697-706
30. Seitz G, Kampchen T, Overheu W. Inverse Diels-Alder Additions,.8. Azepines As Inverse Dienophiles. Archiv der Pharmazie. 1978;311:786-95
31. Neunhoeffer H. Hetarenes IV Six-Membered Rings and Larger Hetero-Rings with Maximum Unsaturation. In: (ed.) Gilchchrist L. Houben-Weyl Methods in Organic Chemistry. New York: Thieme Verlag. 1998:870-916
32. Massa W, Kang HC, Rischke M. et al. Cycloaddition Reactions of Cyclopropenone with 1,2,4-Triazines, A New Synthesis of Pyrazolo[1,2-A]1,2,4-Triazin-6-Ones. Archiv der Pharmazie. 1994;327:477-80
33. Boger DL, Panek JS. Inverse Electron Demand Diels-Alder Reactions of Heterocyclic Azadienes - Formal Total Synthesis of Streptonigrin. Journal of the American Chemical Society. 1985;107:5745-54
34. Wan ZK, Woo GHC, Snyder JK. Dienophilicity of imidazole in inverse electron demand Diels-Alder reactions: cycloadditions with 1,2,4,5-tetrazines and the structure of zarzissine. Tetrahedron Letters. 2001;57:5497-507
35. Bachmann WE, Deno NC. The Diels-Alder Reaction of 1-Vinylnaphthalene with a,@- and a,p,y,G- Unsaturated Acids and Derivatives. J Americ Chem Soc. 1949;71:362-3
36. Sauer J, Wiest H. Diels-Alder-Additionen Mit Inversem Elektronenbedarf. Angewandte Chemie-International Edition. 1962;74:353
37. Pipkorn R, Waldeck W, Didinger B. et al. Inverse-electron-demand Diels-Alder reaction as a highly efficient chemoselective ligation procedure: Synthesis and function of a BioShuttle for temozolomide transport into prostate cancer cells. J Pept Sci. 2009;15:235-41
38. Waldeck W, Wiessler M, Ehemann V. et al. TMZ-BioShuttle--a reformulated temozolomide. Int J Med Sci. 2008;5:273-84
39. Braun K, Wiessler M, Ehemann V. et al. Treatment of glioblastoma multiforme cells with temozolomide-BioShuttle ligated by the inverse Diels-Alder ligation chemistry. Drug Design, Development and Therapy. 2008;2:289-301
40. Curtius T, Darapsky A, Muller E. Über die Umwandlungsprodukte des Diazoessigesters unter dem Einfluss von Alkalien. Chem Ber. 1908;41:3161-72
41. Boger DL, Coleman RS, Panek JS. et al. A detailed, convenient preparation of dimethyl 1,2,4,5-tetrazine-3,6-dicarboxylate. J Org Chem. 1985;50:5377-9
42. Avram M, Dinmulescu IG, Marica E. et al. Dihydropyridazine aus Olefinen und 3.6-Dicarbomethoxy-1.2.4.5-tetrazin. Chem Ber. 1962;95:2248-53
43. Kampchen T, Massa W, Overheu W. et al. Reactions of Dimethyl "1,2,4,5-Tetrazine-3,6-Dicarboxylate with Nucleophiles. Chem Ber. 1982;115:683-94
44. Chang YS, Jeong JM, Lee YS. et al. Preparation of 18F-human serum albumin: a simple and efficient protein labeling method with 18F using a hydrazone-formation method. Bioconjug Chem. 2005;16:1329-33
45. Sauer J, Heinrich G. Kinetik und Umsetzungen Von 1.2.4.5-Tetrazinen Mit Winkelgespannten und Elektronenreichen Doppelbindungen. Tetrahedron Letters. 1966:4979
46. Reppe W, Schlichting O, Klager K. et al. Cyclisierende Polymerisation von Acetylen I. Justus Liebigs Annalen der Chemie. 1948;560:1-92
47. Avram M, Mateescu G, Nenitzescu CD. Die Konfiguration der Addukte des Cyclooctatetraens Mit Maleinsaureanhydrid und Acetylendicarbonsaure-Dimethylester. Annalen der Chemie-Justus Liebig. 1960;636:174-83
48. Huisgen R, Mietzsch F. Valence Tautomerism of Cyclooctatetraene. Angewandte Chemie-International Edition. 1964;3:83
49. Vogel E. Kleine Kohlenstoff-Ringe.2. Uber Die Thermische Stabilitat des Kohlenstoff-Vierrings. Annalen der Chemie-Justus Liebig. 1958;615:14-21
50. Hartmann W. Photosensitized Addition of Maleic Anhydride to Terminal Alkynes. Chemische Berichte-Recueil. 1969;102:3974
Author contact
![]() Correspondence to: Dr. Klaus Braun, German Cancer Research Center (DKFZ), Dept. of Imaging and Radiooncology, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany. Tel: +49 6221 42 2495; Fax: +49 6221 42 3326. k.braunde
Correspondence to: Dr. Klaus Braun, German Cancer Research Center (DKFZ), Dept. of Imaging and Radiooncology, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany. Tel: +49 6221 42 2495; Fax: +49 6221 42 3326. k.braunde