Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2009; 6(2):93-101. doi:10.7150/ijms.6.93 This issue Cite
Research Paper
PKC and PKA Phosphorylation Affect the Subcellular Localization of Claudin-1 in Melanoma Cells
Amanda D. French1, Jennifer L. Fiori1 #, Tura C. Camilli1, Poloko D. Leotlela1, Michael P. O'Connell1, Brittany P. Frank2, Sarah Subaran2, Fred. E. Indig2, Dennis D. Taub1, Ashani T. Weeraratna1 ![]()
1. Laboratory of Immunology, National Institute on Aging, Baltimore, MD 21124, USA
2. Research Resources Branch, National Institute on Aging, Baltimore, MD 21124, USA
# Present Address: Laboratory of Clinical Investigation, National Institute on Aging, Baltimore, MD 21124, USA
Received 2009-2-27; Accepted 2009-3-12; Published 2009-3-12
Abstract
Cytoplasmic expression of claudin-1 in metastatic melanoma cells correlates to increased migration, and increased secretion of MMP-2 in a PKC dependent manner, whereas claudin-1 nuclear expression is found in benign nevi. Melanoma cells were transfected with a vector expressing CLDN-1 fused to a nuclear localization signal (NLS). Despite significant nuclear localization of claudin-1, there was still transport of claudin-1 to the cytoplasm. Phorbol ester treatment of cells transfected with NLS-claudin-1 resulted in an exclusion of claudin-1 from the nucleus, despite the NLS. To ascertain whether PKC or PKA were involved in this translocation, we mutated the putative phosphorylation sites within the protein. We found that mutating the PKC phosphorylation sites to mimic a non-phosphorylated state did not cause a shift of claudin-1 to the nucleus of the cells, but mutating the PKA sites did. Mutations of either site to mimic constitutive phosphorylation resulted in cytoplasmic claudin-1 expression. Stable claudin-1 transfectants containing non-phosphorylatable PKA sites exhibited decreased motility. These data imply that subcellular localization of claudin-1 can be controlled by phosphorylation, dicating effects on metastatic capacity.
Keywords: Claudin, melanoma, metastasis, PKC, PKA
Introduction
Claudin-1 is a member the claudin family of proteins, which are important in tight junction formation (1). Tight junction proteins are located along the cell membrane at the apical edges. They play roles in major cellular functions such as growth and adhesion and are responsible for regulating the paracellular transport of molecules (2). Claudins were first shown to be abnormally expressed in breast and ovarian cancer (3), and have since been found to play roles in other cancers such as melanoma (4), renal and squamous cell carcinomas (2), to name just a few. Because claudins regulate paracellular transport, they are usually found at the cell membrane. However, these proteins, and other tight junction proteins such as zona occludens-1 (ZO-1) have been shown to alter their subcellular localization during malignant progression (5). In melanocytic lesions, we have shown that claudin-1 expression is not only increased, but that its subcellular localization becomes dysregulated, moving away from its typical location at the cell membrane (4). Benign lesions and less aggressive melanomas express claudin-1 in the nucleus, whereas aggressive melanomas have an abundance of claudin-1 in the cytoplasm. We have shown that increasing claudin-1 expression in less metastatic melanoma cells caused an increase in melanoma cell motility, as well as in levels of MMP-2 activation and secretion. These effects could be reversed using siRNA directed against claudin-1. Increased claudin-1 expression did not correlate to increases in tight junction function, presumably due to the fact that it was expressed in the cytoplasm, rather than solely at the cell membrane (4).
Other studies have shown that protein kinase activity is important for the regulation and localization of claudin expression. It has been shown that protein kinases such as Protein Kinase A (PKA) and Protein Kinase C (PKC) can phosphorylate claudins (6,7). Phorbol ester treatment, which activates conventional PKC isoforoms, increased the expression (8) and cytoplasmic distribution of claudin-1, and simultaneously reduced tight junction barrier integrity (7). Conversely, activation of atypical PKC isoforms using bryostatin instead increased the expression of claudin-1 in the tight junction complex, and increased tight junction barrier integrity (9). In melanoma, we have shown that claudin-1 expression levels are increased upon phorbol ester treatment, and decreased by inhibitors of conventional PKC isoforms, implying that, in melanoma, claudin-1 expression is dependent upon PKC isoforoms such as α, β and γ (4). In this study, we also examine the effects of PKA on claudin-1 expression and localization, and how the localization of claudin-1 can affect melanoma cell motility.
Significance
Tight junction proteins have long been thought to be solely responsible for the transport of paracellular molecules. Recent data from cancer studies indicate that these proteins also play important roles in signal transduction. In part, this is facilitated by the translocation of claudins to subcellular locations other than their “normal” location at the cell membrane. We show here that phosphorylation modifications of the tight junction protein claudin-1 cause its translocation to the cytoplasm and nucleus and that the subcellular localization of claudin-1 may dictate the metastatic capacity of melanoma cells. Our findings suggest that nuclear versus cytoplasmic expression of claudin-1 may become a valuable marker for diagnosis of malignant melanoma.
Results
PKC activation increases export of claudin-1 from the nucleus
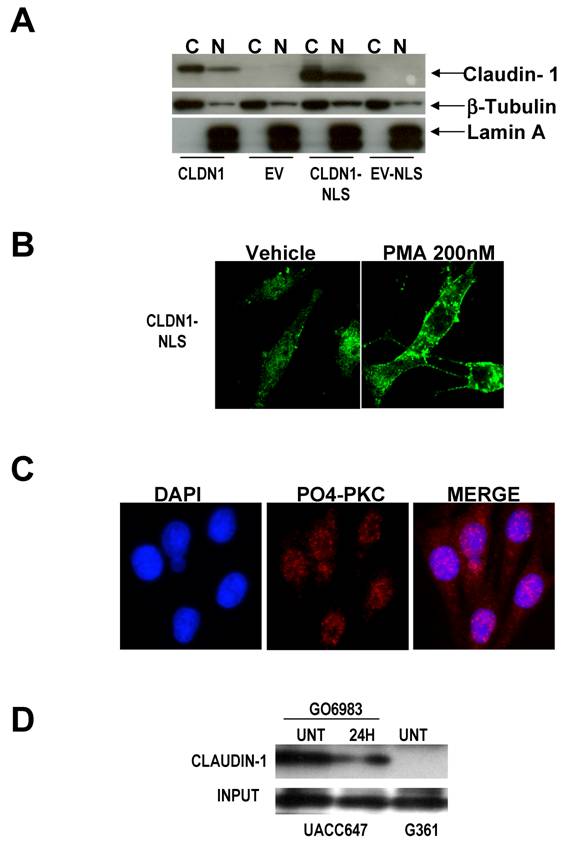
Nuclear localization of claudin-1 is evident in nevi, and less metastatic melanoma cells (4). To determine if the nuclear localization of claudin-1 was related to the increased invasive capacity of melanoma cells, we created a claudin-1 expression vector expressing claudin-1 containing a nuclear localization signal (pDsRedCLDN1-NLS). However, transfection of this vector into G361 melanoma cells (which have low levels of claudin-1) resulted in both nuclear and cytoplasmic expression of claudin-1 (Figure 1 A) despite the fact that claudin-1 was attached to a NLS. This led us to ask whether a post-translational modification such as phosphorylation might be resulting in the transport of claudin-1 out of the nucleus. Since we have previously implicated PKC in melanoma progression, and claudin-1 expression, we transfected pDsRedCLDN1-NLS into G361 cells, and then treated the transfected cells with phorbol ester (PMA). Treatment of cells with 200nM PMA resulted in an almost complete exclusion of claudin-1 from the nucleus (Figure 1B). This implies that active PKC may exist in the nucleus of melanoma cells, and the presence of active PKC isoforms in the nuclei of many cell types has been confirmed (10-12). Staining of the G361 cells with antibodies to phosphorylated PKC (α, β, γ) demonstrates that there is active PKC in melanoma cell nuclei as well (Figure 1C). Further, we performed immunoprecipitation studies using an antibody that binds to any protein that is a potential PKC substrate, followed by western analysis for claudin-1. In UACC647 melanoma cells which are highly metastatic and have high levels of claudin-1 (4), claudin-1 co-immunoprecipitates with the PKC substrate antibody (Figure 1D). In the presence of PKC inhibitors, which we have previously demonstrated to decrease claudin-1 expression (4), there is a decrease in the levels of claudin-1 precipitated by the PKC substrate antibody. G361 cells, which have very little endogenous claudin-1, and low levels of PKC, show no immunoprecipitation of claudin-1 with the PKC substrate antibody (Figure 1D). These data indicated that claudin-1 is a likely target for PKC phosphorylation.
Claudin-1 is not expressed solely in the nucleus even when under the direction of a nuclear localization signal. A) Cytoplasmic and nuclear extracts of G361 melanoma cells (low claudin-1 expressers) were transfected with wild type CLDN1 (CLDN1). By Western analysis, these transfectants demonstrate the presence of claudin-1 protein in the cytoplasm, with small amounts in the nucleus. When cells are transfected with CLDN1 under the control of a nuclear localization signal (CLDN1-NLS), there is increased expression of claudin-1 in the nucleus, but there are still large amounts of claudin-1 in the cytoplasm as well. B) All of the claudin-1 in the nucleus can be shuttled into the cytoplasm by treatment with the PKC activator PMA (phorbol ester). C) This implies that active PKC may exist in the nucleus of melanoma cells, so cells were stained with an antibody to pan-PO4-PKC. Confocal microscopy demonstrates that active PKC can be found in the nucleus of G361 melanoma cells. D) To determine if claudin-1 was a potential PKC substrate, cell lysates from claudin-1 high UACC647 cells, and claudin-1 low G361 cells were subjected to immunoprecipitation using a PKC substrate antibody, and western analysis for claudin-1 was performed. GO6983, a PKC inhibitor, decreases the amount of claudin-1 that is immunoprecipitated by the PKC substrate antibody. G361 cells have very little claudin-1 and thus, none is precipitated by the PKC substrate antibody, indicating the specificity of this immunoprecipitation.
PKC and PKA play distinct roles in the expression and localization of claudin-1
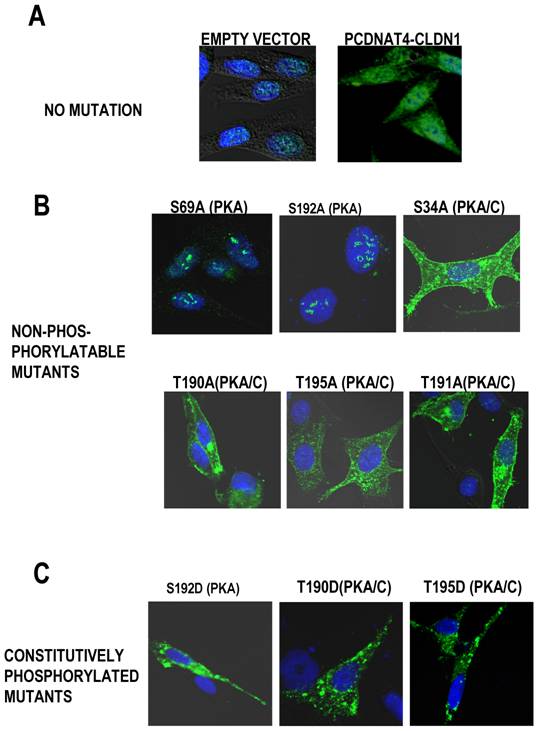
To better characterize the phosphorylation of claudin-1 by PKC, we performed an in silico analysis of the putative PKC phosphorylation sites for claudin-1. We found that there are five putative PKC phosphorylation sites on the claudin-1 protein (212 amino acids long), but all of these are also sites of PKA phosphorylation (Table 1). There are however, three unique sites of PKA phosphorylation, at amino acid residues 65-69, and 189-192 and 201-205. It should be noted that the aa189-192 site overlaps with a PKC/PKA phosphorylation site (aa188-191). To assess whether PKC or PKA was responsible for the phosphorylation and nuclear exclusion of claudin-1, we used site-directed mutagenesis of our PCDNA3.1-CLDN1 vector to make mutants that either mimicked a constitutively phosphorylated state (conversion of the phosphorylation site to an aspartic acid residue, referred to as “D” mutants) or mutants that are non-phosphorylable (conversion of the phosphorylation site to an alanine residue, referred to as “A” mutants). This vector, when transfected into melanoma cells shows both cytoplasmic and nuclear expression of claudin-1, as compared to empty vector controls (Figure 2A). The sites of phosphorylation close to the end of the protein sequence (aa200-212) did not provide us with successful mutants. Mutations to alanine in PKC/PKA sites did not affect the subcellular localization of claudin-1, but alanine mutations of the two unique PKA sites caused nuclear localization of claudin-1 (Figure 2B). Mutations to aspartic acid in both PKA only as well as PKC/PKA sites caused cytoplasmic redistribution of claudin-1 with complete exclusion from the nucleus (Figure 2C).
Site-directed mutagenesis. Putative sites of PKC and PKA phosphorylation on the claudin-1 protein, and the primers used to perform site-directed mutagenesis of these sites. The first 10 rows represent mutations to alanine, and the last ten rows represent mutations to aspartic acid.
| AMINO ACID SEQUENCE | MOTIF (PO4) | PKA/PKC | DNA BASE CHANGE | PRIMER | |
|---|---|---|---|---|---|
| 31-34 | RIYS | RXXpS | PKA | T100G | F: 5'-cagtggaggatttacgcctatgccggcgaca-3' R: 5'-tgtcgccggcataggcgtaaatcctccactg-3' |
| 31-34 | RIYS | [R/K]XX[pS/pT] | PKC | T100G | |
| 65-69 | KVFDS* | KXX[pS/pT] | PKA | T205G | F: 5'-agtgcaaagtctttgacgccttgctgaatctgagc-3' R: 5'-gctcagattcagcaaggcgtcaaagactttgcact-3' |
| 188-190 | RKT | [R/K]X[pS/pT] | PKA | A568T | F: 5'-tgttcctgtccccgaaaatcaacctcttacccaac-3' R: 5'-gttgggtaagaggttgattttcggggacaggaaca-3' |
| 188-190 | RKT | [R/K]X[pS/pT] | PKC | A568T | |
| 188-191 | RKTT | [R/K]XX[pS/pT] | PKC | A571G | F:5'-gctgttcctgtccccgaaaaacagcctcttacccaa-3' R:5'-ttgggtaagaggctgtttttcggggacaggaacagc-3' |
| 188-191 | RKTT | [R/K][R/K]X[pS/pT] | PKA | A571G | |
| 189-192 | KTTS* | KXX[pS/pT] | PKA | A568T_A571G | F:5'-ctgttcctgtccccgaaaatcagcctcttacccaacac-3' R:5'-gtgttgggtaagaggctgattttcggggacaggaacag-3' |
| 195-197 | TPR | [pS/pT]X[R/K] | PKA | A589G_G590C | F:5'-aacaacctcttacccaacaccagcgccctatccaaaacc-3' R:5'-ggttttggatagggcgctggtgttgggtaagaggttgtt-3' |
| 195-197 | TPR | [pS/pT]X[R/K] | PKC | A589G_G590C | |
| 31-34 | RIYS | RXXpS | PKA | T100G_C102A | F:5'-gccccagtggaggatttacgcatatgccggcgaca-3' R:5'-tgtcgccggcatatgcgtaaatcctccactggggc-3' |
| 31-34 | RIYS | [R/K]XX[pS/pT] | PKC | T100G_C102A | |
| 65-69 | KVFDS | KXX[pS/pT] | PKA | T205G_C206A | F:5'-ccagtgcaaagtctttgacgacttgctgaatctgagcagc-3' R:5'-gctgctcagattcagcaagtcgtcaaagactttgcactgg-3' |
| 188-190 | RKT | [R/K]X[pS/pT] | PKA | A568G_C569A_A570C | F:5'-ctttgctgttcctgtccccgaaaagacacctcttacccaacacca-3' R:5'-tggtgttgggtaagaggtgtcttttcggggacaggaacagcaaag-3' |
| 188-190 | RKT | [R/K]X[pS/pT] | PKC | A568G_C569A_A570C | |
| 188-191 | RKTT | [R/K]XX[pS/pT] | PKC | A571G_C572A | F:5'-ttcctgtccccgaaaaacagactcttacccaacaccaagg-3' R:5'-ccttggtgttgggtaagagtctgtttttcggggacaggaa-3' |
| 188-191 | RKTT | [R/K][R/K]X[pS/pT] | PKA | A571G_C572A | |
| 189-192 | KTTS | KXX[pS/pT] | PKA | A568G_C569A_A570C_A571G_C572A | F:5'-actttgctgttcctgtccccgaaaagacgactcttacccaacaccaaggccc-3' R:5'-gggccttggtgttgggtaagagtcgtcttttcggggacaggaacagcaaagt-3' |
| 195-197 | TPR | [pS/pT]X[R/K] | PKA | A589G_G590A_G591C | F:5'-aaaacaacctcttacccaacaccagacccctatccaaaacctgca-3' R:5'-tgcaggttttggataggggtctggtgttgggtaagaggttgtttt-3' |
| 195-197 | TPR | [pS/pT]X[R/K] | PKC | A589G_G590A_G591C | |
Site-directed mutagenesis of PKA/PKC in CLDN1. A) G361 melanoma cells which have very little endogenous claudin-1 were transfected with either an empty vector or CLDN-1 and localization was observed using confocal microscopy. Site directed mutagenesis was performed to render the potential sites of PKC and PKA phosphorylation on the claudin-1 protein non phosphorylatable or to mimic constitutive activation of PKC or PKA. B) Rendering the serine at position 69 on the claudin-1 protein non-phosphorylatable causes nuclear localization of claudin-1. C) Mutations mimicking constitutive activation of its phosphorylatable sites cause exclusively cytoplasmic localizationof claudin-1.
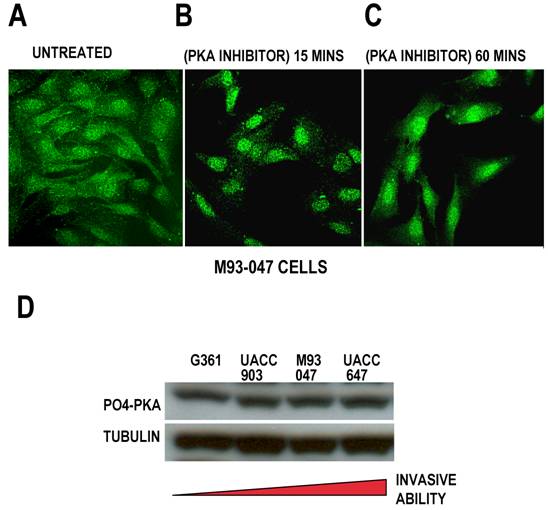
Nuclear localization of claudin-1 upon mutation of the PKA sites could be mimicked using PKA inhibitors. Untreated M93-047 cells have high levels of claudin-1 and exhibit a diffuse, largely cytoplasmic pattern of claudin-1 (Figure 3A). Upon treatment with PKA inhibitors for 15 minutes, claudin-1 shuttles into the nucleus (Figure 3B). After 1 hour of treatment with PKA inhibitor, claudin-1 is still highly nuclear, although some claudin-1 has started to shuttle out of the nucleus (Figure 3C). This is unlike the situation with PKC inhibitors, which cause a general downregulation of claudin-1 expression (4). Furthermore, it is interesting to note that all of the cell lines have similar levels of phospho-PKA (Figure 3D) which explains why the transfected claudin-1 is shuttled out of the nucleus even when transfected into less metastatic G361 cells, which have low amounts of phospho-PKC (4,13). Taken together these data indicate that PKA is likely contributing to the subcellular localization of claudin-1.
Pharmacological deactivation of PKA also causes nuclear translocation of claudin-1. A) Claudin-1 high M93-047 cells have abundant expression of claudin-1 in the cytoplasm, with some nuclear expression of claudin-1. B) Treatment of these cells with PKA inhibitor results in nuclear translocation of claudin-1 in as little as 15 minutes, with C) sustained nuclear localization at 1 hour. D) All melanoma cell lines, regardless of claudin-1 status, or metastatic ability have similar levels of active PKA.
Nuclear claudin-1 does not increase melanoma cell motility
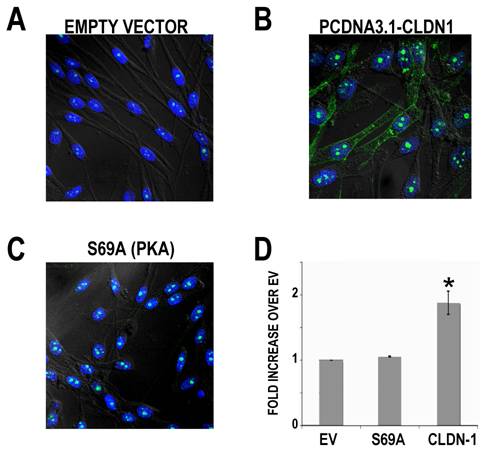
We have previously shown that increasing the levels of CLDN-1 increases the invasion of melanoma cells (4). To determine if the increase in claudin-1 needs to be cytoplasmic and not nuclear to affect the ability of melanoma cells to invade, we first performed a stable transfection of our G361 cells with the S69A (PKA, non-phosphorylatable) claudin-1 mutants. Pooled stable clones were analyzed for the expression of claudin-1 and its subcellular localization. As can be seen the PCDNA3.1-CLDN1 transfected cells have plenty of cytoplasmic claudin as compared to the stable empty vector clones (Figure 4A, B) whereas the S69A mutants have largely nuclear expression of claudin-1 (Figure 4C). To test their invasive capacity, stable clones were allowed to invade through a Matrigel-coated invasion chamber. As compared to empty vector controls, cells transfected with the PKA deactivating S69A mutation did not show any increase in invasion. However, G361 cells transfected with the claudin-1 overexpressing vector showed a nearly 2-fold increase in invasion as compared to the empty vector control (Figure 4D). These data appear to support the hypothesis that nuclear overexpression of claudin-1 does not increase the invasive capacity of melanoma cells, where cytoplasmic expression of claudin-1 does.
Nuclear claudin-1 cannot increase the invasive ability of G361 melanoma cells. Stable transfectants of the empty vector (A), the PCDNA3.1-CLDN1 vector (B), and the PCDNA3.1-CLDN1 vector containing the S69A mutation (C) were created. These cells were then subjected to invasion through Matrigel in a modified Boyden chamber assay. Only the PCDNA3.1-CLDN1 stable transfectants demonstrated an increase in invasion (D).
Discussion
Claudin-1 has been shown to play an important role in skin development and tumorigenesis. Claudin-1 null mice die from dehydration within a few hours of birth due to the breakdown of barrier integrity in the epidermis (14). In humans, patients with the pathological condition known as AEC (Ankyloblepharon-Ectodermal dysplasia-Clefting) also have compromised skin integrity, and this has been shown to correlate to the lack of claudin-1 and also to the loss of p63 (15). Further, we have previously shown that increasing claudin-1 expression increases melanoma metastasis (4). In this study we demonstrate that the nuclear localization of claudin-1 cannot contribute to the migratory capacity of melanoma cells. These data confirm our previous observations that nuclear claudin-1 is expressed largely in benign nevi, and early stage melanoma, but that highly metastatic melanoma cells tend to have increased claudin-1 in the cytoplasm and at the membrane, but very little nuclear staining (4). Further, we demonstrate that claudin-1 subcellular localization is affected by PKA, although constitutively activating both PKA and PKC sites results in the cytoplasmic distribution of claudin-1. However, the deactivation of PKC sites does not cause nuclear localization of claudin-1. Unlike PKC, which we have previously shown to decrease the levels of claudin-1 expression, PKA inhibition does not appear to affect claudin-1 levels, but does cause nuclear sequestration of claudin-1, implying that PKA is required for claudin-1 exclusion from the nucleus. It is curious that in cases where claudin-1 is low, it is predominantly nuclear, despite abundant active PKA. This observation implies that the levels of claudin-1 may need to reach a certain threshold prior to being shuttled out of the nucleus. Hence, in metastatic melanoma cells that have high levels of PKC, there is increased claudin-1 expression, and when enough is expressed in the nucleus, PKA causes its translocation to the cytoplasm.
It is unclear what the significance of claudin-1 in the nucleus may be. It is known that the nuclear expression of other tight junction proteins such as ZO-1 can inhibit proliferation (16), but in our stable S69A transfectants, we see no difference in the rate of proliferation between the transfectants with nuclear claudin-1 and the empty vector controls (data not shown). It has been suggested that the translocation of claudin-1 into the nucleus of cells, despite the absence of a nuclear localization signal may require APC, ZO-1 or ZO-2, possibly as shuttles for claudin-1 (17). This opens up the possibility that Wnt signaling is involved in claudin-1 expression and localization. Indeed, a study shows that increasing β-catenin increases the expression of claudin-1 in colorectal cancer cells, and that claudin-1 expression is increased in more aggressive cancers (18). We have previously demonstrated the importance of the non-canonical Wnt signaling pathway in melanoma (13,19,20), and have shown that Wnt5A increases the epithelial to mesenchymal transition (EMT) in melanoma cells, leading to increased invasion (13). These effects all require PKC, and since we have shown that, in melanoma, claudin-1 expression is dependent upon PKC, it is likely that Wnt5A may also increase claudin-1 expression. Studies to confirm this are underway in our laboratory. Further, claudin-1 is important in mediating an EMT in colon cancer cells (17), similar to that seen with Wnt5A. All of these data taken together imply that claudin-1 is an important marker of melanoma progression, especially when its subcellular localization is considered, as we have shown that its cytoplasmic expression is critical for increased malignancy. How it interacts with other molecules involved in melanoma malignancy, such as members of the Wnt pathway, remains to be determined.
Materials and Methods
Cell lines. As previously described (4), the human melanoma cell lines UACC647 and M93-047 were cultured in RPMI-1640 media (Gibco-BRL, Bethesda, MD) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT), 100 units/ml Penicillin G and 100 units streptomycin (Gibco-BRL). G361 cells were maintained in McCoy's 5A medium (Gibco-BRL) with 10% FBS. All cell cultures were incubated at 37°C in 5% CO2.
Claudin-1 expression vectors and site-directed mutagenesis. CLDN1 was cloned into mammalian expression vector pcDNA3.1/V5-His© as previously described (4). Site-directed mutagenesis was performed on the claudin-1 sequence in the pcDNA3.1/V5-His vector using the Quikchange II site-directed mutagenesis kit (Stratagene, La Jolla, CA) to mutate the sites indicated in Table 1. For initial experiments attempting to attach CLDN1 to a nuclear localization signal, CLDN1 was then subcloned into the PdsRed2-Nuc vector (Clontech, Mountainview, CA). The correct sequence and orientation of all constructs was verified by direct DNA sequencing.
Transfections. Cells were allowed to reach 60-80% confluency within 48 hours of seeding. The cells were transfected with claudin-1 expression vectors or empty vector controls, using Lipofectamine Plus (Gibco-BRL). After six hours of transfection, the medium was replaced with fresh serum-containing medium. For stable transfections, cells were treated with 500μg/mL G418, clones were selected and analyzed for CLDN1 expression by confocal microscopy using a Zeiss Meta 510 confocal microscope (Zeiss, Thornwood, NY).
Treatments. For PKC inhibition studies Gö 6983 (Sigma, St. Louis, MO) was used at a concentration of 1uM, and PMA (Cell Signaling, Beverly, MA) was used at a concentration of 200nM as previously described (4). For vehicle controls, cells were treated with equivalent amounts of DMSO. PKA inhibitor (cell permeable, myristoylated Protein Kinase A Inhibitor Amide 14-22, Calbiochem, San Diego, CA), was used at a concentration of 10μM for the indicated times. The PKA activator, forskolin (Sigma) was used at a concentration of 5 μM for the indicated times.
Immunofluorescenct confocal microscopy. As previously described (4), for claudin-1 staining, cells were grown on glass slides, and allowed to reach 80% confluency. Cells were fixed using 95% methanol, washed and blocked with a fluorescent blocking buffer (0.2% Triton-X100, 0.2% BSA, 0.2%, Casein, 5% normal goat serum, 0.2% gelatin, 0.02% sodium azide) for 1 hour at room temperature. Incubation in claudin-1 primary antibody at 0.25μg/ml (ZYMED Laboratories, San Francisco, CA) was performed overnight at 4oC. The cells were washed again with PBS for 30 minutes, probed with secondary antibody (conjugated with Alexa fluor-488, Invitrogen, Carlsbad, CA), then washed again with PBS, mounted in Pro-long Gold® (Invitrogen, Carlsbad, CA) with DAPI and then examined using confocal microscopy. Images were taken using a Zeiss Meta 510 confocal microscope.
Western Analysis. β-tubulin (1:1000), Lamin-A (1:1000), Phospho- PKA (1:1000) and PKC substrate (1:200) antibodies were obtained from Cell Signaling (Beverly, MA). Claudin-1 antibody (1:1000) was obtained from Zymed Laboratories (San Francisco, CA). Cells were grown to 80% confluency and then harvested on ice as previously described (4). Nuclear and cytoplasmic extracts were obtained according to manufacturer's instructions using the NE-PER extraction kit (Pierce, Rockford, IL). Proteins were quantitated using a Pierce BCA protein quantitation assay (Pierce). 50μg of each lysate was run out on SDS-PAGE 10% Tris-glycine Nu-PAGE gels (Invitrogen) and transferred onto 0.2 micron PVDF. The membranes were probed with antibodies and then visualized using the ECL system (Amersham Biosceinces, Piscataway, NJ).
For immunoprecipitation, 250μg of lysate was incubated with protein A/G agarose (Sigma) for 4 hours to clear the lysate. The mixture was centrifuged gently, and the lysate was removed from the beads and placed into a new, siliconized microfuge tube. PKC substrate antibody was added at a ratio of 1:200, and the lysate and antibody were incubated with gentle rocking overnight at 4°C. Protein A/G agarose was then added for 6h, after which time the lysate was removed, and the agarose washed in RIPA three times. SDS-based sample buffer was used to remove the bound proteins from the agarose, and the entire mixture was subjected to Western analysis for claudin-1 as described above.
Invasion Assays. Matrigel invasion assays were performed using transwell migration chambers (Corning life Sciences, Lowell, MA) as previously described (21). Briefly, 8μM filters were coated with 150μL of 80μg/mL reconstituted basement membrane (Matrigel®, Becton Dickinson, Franklin Lakes, NJ). Wild type or mutant claudin-1 expressing clones were serum starved for 16h, and 2 X 105 cells were seeded onto the filters. The total volume on the top of the filter was adjusted to 800μL of serum free medium and 800μL of medium containing 20% fetal calf serum was placed in the lower well to act as a chemoattractant. Cells were allowed to invade and adhere to the lower chamber, after which they were stained using crystal violet, and counted. All cell lines were assayed in triplicate.
Statistics. A Student's t-test was performed when required in at least 3 independent experiments. Significance was designated as *p<0.05, **p<0.01.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute on Aging. We thank Dr. Pat J. Morin for helpful comments on the manuscript.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Heiskala M, Peterson P.A, Yang Y. The roles of claudin superfamily proteins in paracellular transport. Traffic. 2001;2(2):93-98
2. Morin P.J. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005;65(21):9603-9606
3. Hough C.D, Sherman-Baust C.A, Pizer E.S, Montz F.J, Im D.D, Rosenshein N.B, Cho K.R, Riggins G.J, Morin P.J. Large-scale serial analysis of gene expression reveals genes differentially expressed in ovarian cancer. Cancer Res. 2000;60(22):6281-6287
4. Leotlela P.D, Wade M.S, Duray P.H, Rhode M.J, Brown H.F, Rosenthal D.T, Dissanayake S.K, Earley R, Indig F.E, Nickoloff B.J, Taub D.D, Kallioniemi O.P, Meltzer P, Morin P.J, Weeraratna A.T. Claudin-1 overexpression in melanoma is regulated by PKC and contributes to melanoma cell motility. Oncogene. 2007;26(26):3846-3856
5. Smalley K.S, Brafford P, Haass N.K, Brandner J.M, Brown E, Herlyn M. Up-regulated expression of zonula occludens protein-1 in human melanoma associates with N-cadherin and contributes to invasion and adhesion. Am J Pathol. 2005;166(5):1541-1554
6. D'Souza T, Agarwal R, Morin P.J. Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem. 2005;280(28):26233-26240
7. Lippoldt A, Liebner S, Andbjer B, Kalbacher H, Wolburg H, Haller H, Fuxe K. Organization of choroid plexus epithelial and endothelial cell tight junctions and regulation of claudin-1, -2 and -5 expression by protein kinase C. Neuroreport. 2000;11(7):1427-1431
8. Koizumi J, Kojima T, Ogasawara N, Kamekura R, Kurose M, Go M, Harimaya A, Murata M, Osanai M, Chiba H, Himi T, Sawada N. Protein kinase C enhances tight junction barrier function of human nasal epithelial cells in primary culture by transcriptional regulation. Molecular pharmacology. 2008;74(2):432-442
9. Yoo J, Nichols A, Mammen J, Calvo I, Song J.C, Worrell R.T, Matlin K, Matthews J.B. Bryostatin-1 enhances barrier function in T84 epithelia through PKC-dependent regulation of tight junction proteins. American journal of physiology. 2003;285(2):C300-309
10. Marchisio M, Bertagnolo V, Lanuti P, Gaspari A.R, Paludi M, Ciccocioppo F, Ercolino E, Bascelli A, Cataldi A, Miscia S. Nuclear protein kinase C-delta: a possible check-point of cell cycle progression. International journal of immunopathology and pharmacology. 2006;19(2):287-291
11. Kenessey A, Sullivan E.A, Ojamaa K. Nuclear localization of protein kinase C-alpha induces thyroid hormone receptor-alpha1 expression in the cardiomyocyte. Am J Physiol Heart Circ Physiol. 2006;290(1):H381-389
12. Hu T, Exton J.H. Protein kinase Calpha translocates to the perinuclear region to activate phospholipase D1. J Biol Chem. 2004;279(34):35702-35708
13. Dissanayake S.K, Wade M, Johnson C.E, O'Connell M.P, Leotlela P.D, French A.D, Shah K.V, Hewitt K.J, Rosen-thal D.T, Indig F.E, Jiang Y, Nickoloff B.J, Taub D.D, Trent J.M, Moon R.T, Bittner M, Weeraratna A.T. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem. 2007;282(23):17259-17271
14. Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156(6):1099-1111
15. Lopardo T, Lo Iacono N, Marinari B, Giustizieri M.L, Cyr D.G, Merlo G, Crosti F, Costanzo A, Guerrini L. Claudin-1 is a p63 target gene with a crucial role in epithelial development. PLoS ONE. 2008;3(7):e2715
16. Traweger A, Fuchs R, Krizbai I.A, Weiger T.M, Bauer H.C, Bauer H. The tight junction protein ZO-2 localizes to the nucleus and interacts with the heterogeneous nuclear ribonucleoprotein scaffold attachment factor-B. J Biol Chem. 2003;278(4):2692-2700
17. Dhawan P, Singh A.B, Deane N.G, No Y, Shiou S.R, Schmidt C, Neff J, Washington M.K, Beauchamp R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J Clin Invest. 2005;115(7):1765-1776
18. Miwa N, Furuse M, Tsukita S, Niikawa N, Nakamura Y, Furukawa Y. Involvement of claudin-1 in the beta-catenin/Tcf signaling pathway and its frequent upregulation in human colorectal cancers. Oncol Res. 2001;12(11-12):469-476
19. Dissanayake S.K, Olkhanud P.B, O'Connell M.P, Carter A, French A.D, Camilli T.C, Emeche C.D, Hewitt K.J, Rosenthal D.T, Leotlela P.D, Wade M.S, Yang S.W, Brant L, Nickoloff B.J, Messina J.L, Biragyn A, Hoek K.S, Taub D.D, Longo D.L, Sondak V.K, Hewitt S.M, Weeraratna A.T. Wnt5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res. 2008;68(24):10205-10214
20. Weeraratna A.T, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent J.M. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002;1(3):279-288
21. O'Connell M.P, French A.D, Leotlela P.D, Weeraratna A.T. Assaying Wnt5A-mediated invasion in melanoma cells. Methods in molecular biology (Clifton NJ). 2008;468:243-253
Author contact
![]() Correspondence to: Ashani T. Weeraratna, PhD, Laboratory of Immunology, National Institutes of Health, National Institute on Aging, Biomedical Research Center, 251 Bayview Blvd, RM 08C226, Baltimore, Maryland 21224. Voice: (410) 558-8146; Fax: (410) 558-8284; Email: weeraratnia.nih.gov
Correspondence to: Ashani T. Weeraratna, PhD, Laboratory of Immunology, National Institutes of Health, National Institute on Aging, Biomedical Research Center, 251 Bayview Blvd, RM 08C226, Baltimore, Maryland 21224. Voice: (410) 558-8146; Fax: (410) 558-8284; Email: weeraratnia.nih.gov