Impact Factor ISSN: 1449-1907
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results and Discussion
Summary
Material and Methods
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2008; 5(5):230-239. doi:10.7150/ijms.5.230 This issue Cite
Research Paper
HIV-1 Capsid Assembly Inhibitor (CAI) Peptide: Structural Preferences and Delivery into Human Embryonic Lung Cells and Lymphocytes
Klaus Braun1, Martin Frank2, Rüdiger Pipkorn3, Jennifer Reed4, Herbert Spring5, Jürgen Debus6, Bernd Didinger6, Claus-Wilhelm von der Lieth2, Manfred Wiessler1, Waldemar Waldeck7 ![]()
1. German Cancer Research Center, Division of Molecular Toxicology, INF 280, D-69120 Heidelberg, Germany
2. German Cancer Research Center, Division Central Spectroscopy B090, INF 280, D-69120 Heidelberg, Germany
3. German Cancer Research Center, Core Facility Peptide Synthesis, INF 580, D-69120 Heidelberg, Germany
4. German Cancer Research Center, Biomolecular Mechanisms, INF 280, D-69120 Heidelberg, Germany
5. German Cancer Research Center, Research Group Structural Biochemistry, INF 280, D-69120 Heidelberg, Germany
6. University of Heidelberg, Radiation Oncology, INF 110, D-69120 Heidelberg
7. German Cancer Research Center, Division of Biophysics of Macromolecules, INF 580, D-69120 Heidelberg, Germany
Received 2008-6-17; Accepted 2008-7-29; Published 2008-7-31
Abstract
The Human immunodeficiency virus 1 derived capsid assembly inhibitor peptide (HIV-1 CAI-peptide) is a promising lead candidate for anti-HIV drug development. Its drawback, however, is that it cannot permeate cells directly. Here we report the transport of the pharmacologically active CAI-peptide into human lymphocytes and Human Embryonic Lung cells (HEL) using the BioShuttle platform. Generally, the transfer of pharmacologically active substances across membranes, demonstrated by confocal laser scanning microscopy (CLSM), could lead to a loss of function by changing the molecule's structure. Molecular dynamics (MD) simulations and circular dichroism (CD) studies suggest that the CAI-peptide has an intrinsic capacity to form a helical structure, which seems to be critical for the pharmacological effect as revealed by intensive docking calculations and comparison with control peptides. This coupling of the CAI-peptide to a BioShuttle-molecule additionally improved its solubility. Under the conditions described, the HIV-1 CAI peptide was transported into living cells and could be localized in the vicinity of the mitochondria.
Keywords: BioShuttle, Capsid Assembly Inhibitors, Drug Delivery, HIV-Drug Development
Introduction
In the 2007 report of the global AIDS epidemic it was calculated that 30.6 million–36.1 million people world-wide were living with the human immunodeficiency virus (HIV) at the end of 2007. An estimated 1.8–4.1 million became newly infected with HIV and about 1.9–2.4 million people lost their lives by acquired immunodeficiency syndrome (AIDS). In several countries favourable trends in the incidence of AIDS or HIV are related to changes in individual behaviour. Prevention programs raised a slight hope to reduce incidence; however, the epidemics in the world's most affected regions are highly diverse and, especially in Southern Africa and Eastern Europe, still expanding [1]. In addition to the national HIV prevention programs which should promote infection control practices in health-care settings, the development of effective curative therapeutic approaches for HIV-infected patients remains a considerable challenge for both the World Health Organization (WHO) and drug research. Current successful therapies involve the combination of the inhibition of the viral enzyme reverse transcriptase, protease inhibitors and inhibitors of viral entry, described as highly active anti-retroviral therapy (HAART) [2-4]. The striking success of HAART raised hope for the affected people. However, meanwhile, the number of drug-resistant variants of HIV increased and the exploration of new alternative targets is necessary for the next generation of antiviral drug development [5].
The identification of active peptides as attractive candidates for intervention at the virus assembly level is one promising strategy. Briefly, the gag gene produces a 55-kilodalton kD Gag precursor protein (p55 or Pr55Gag), which is expressed from the unspliced viral mRNA [6]. During translation the N-terminus of p55 is myristoylated triggering its association to the cytoplasmic side of cell membranes [7] and the release of the budding of viral particles from the surface of infected cells. After budding, the virus aspartyl protease PR [8] cleaves p55, thus generating a set of smaller proteins and spacer peptides (SP) encoded by the viral pol gene during the process of viral maturation. The proteins are termed: matrix p17 (MA), nucleocapsid p9 (NC), p4 and capsid p24 (CA) and SP1 and SP2 respectively [9]. The assembly of Gag proteins into immature viral particles followed by proteolytic disassembly of the Gag shell to mature capsids are pivotal steps for the formation of infective HIV-1 [10]. The function of CA is of central importance in assembling the conical core of viral particles and so its inhibition is a desirable therapeutic target. Attempts have been made to develop capsid assembly inhibitors (CAI) based on Gag-derived peptide fragments, which are targeted to HIV Gag intermediates. Their intracellular biochemical processes and their mechanism of action in the intervention of the viral life cycle are not yet completely characterized. Molecules like the CAP-1 [11, 12] also termed PA-457 [11, 13, 14] and most notably, the peptide-based CAI(Pep1) [15] are suitable lead compounds for anti HIV drug development. However, these show an insufficient bioavailability due to their limited water solubility. This situation demands intensive efforts for development and characterization of delivery systems capable of transporting sufficient amounts of pharmacologically active agents such as CAI-peptides into the HIV-1 infected cells. In cell-free systems the antiviral activity of CAI-peptides has been documented and the discovery of peptide-based antiviral components is encouraging [15-17]. In this study we describe the synthesis and investigation of the modified peptide-based CAI-BioShuttle delivery platform.
Results and Discussion
It is well documented that the transport efficiency of active substances can depend on the physico-chemical properties of the cargo [18]. In our study we characterized the transmembrane transport and the intracellular fate of the pharmacologically active CAI-probe by confocal laser scanning microscopy (CLSM) in comparison with the respective controls. Constructs harboring the protein transduction domain of HIV-1 Tat(48-60) as a transmembrane transport peptide coupled via an enzymatic cleavable disulfide-bridge to a functional CAI-Inhibitor result in a HIV-1 Tat(48-60)-Cys-S-S-Cys-CAI-conjugate as shown in figure 1. Coupling of such therapeutic CAI-peptides to the modular BioShuttle [19] carrier, could provide effective reduction of viral loads of HIV. In this context the structural modalities of the CAI-peptides such as folding, which are essential for binding at the target site and for the pharmacological effect, remain to be elucidated. Further, their structural behavior after passage across the cellular membranes during their delivery and the structural requirements of their corresponding target sites are still largely unexplored. With in silico methods and CD measurements we could predict the molecular structures of the cargos after passage through membranes and understand better their pharmacological behaviour.
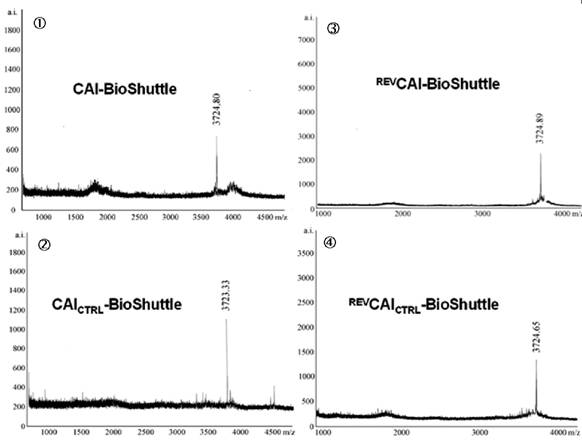
Schematized modular compositions of the CAI-BioShuttle and mass spectra of the investigated conjugates. Top part of the figure: The inhibitor peptide, control peptides, and the transmembrane transport module are connected with a sulfur bridge between the two cysteines (Single letter symbol C [bold]). Horizontally: ① represents the modules of the CAI-BioShuttle, ② the CAICTRL-BioShuttle, and ③ and ④ the BioShuttle connected to the reverse form of the CAI-inhibitor and the control, respectively. Vertically: ① CAI-Inhibitor, ② scrambled control and their corresponding peptides in reverse orientation (③ and ④), respectively. Middle column shows the transmembrane transport module. The link to the RCSB PDB Protein Data Bank is indicated. (4);5)). The corresponding mass spectra of the above listed conjugates are shown at the lower part of the figure.
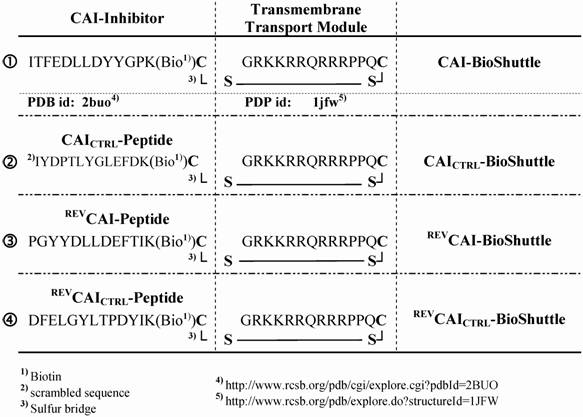
For delivery of the HIV-1 CAI into human cells a bi-modular peptide was developed and constructed consisting of a transport unit for transmembrane transport connected to a peptide with a capsid assembly inhibitory (CAI) effect as a functional unit.
To demonstrate the transport efficiency and to facilitate investigation of both the biochemical and the physico-chemical effects of the CAI, corresponding control peptides were also synthesized. An overview is shown in figure 1;①-④.
Subcellular localization of the CAI-peptides by CLSM
With confocal laser scanning microscopy we could demonstrate the intracellular distribution of the BioShuttle-delivered CAI-peptide (figure 1;①). Parallel to a scrambled control sample [CAICTRL-BioShuttle (figure 1;②)], the corresponding CAI-molecules with reverse peptide sequences [REVCAI-BioShuttle (figure 1;③) and REVCAICTRL-BioShuttle (figure 1;④)] were investigated.
We detected all investigated peptides, namely the CAI-peptide, its control and its corresponding reverse version in the cytoplasm and in nuclei of both peripheral T lymphocytes (PTL) cells and the human embryonic lung (HEL).
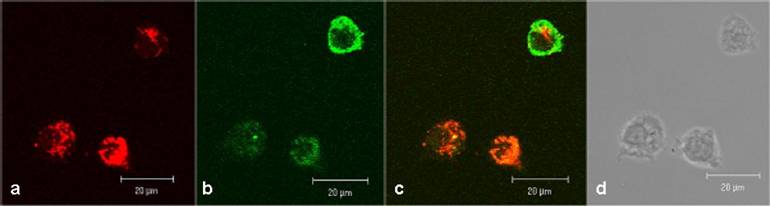
In human lymphocytes, as shown as a DIC picture in figure 2d, we demonstrated by CLSM, strong green fluorescence signals close to cell membrane and distributed in the cytoplasm (figure 2b). Red fluorescence signals (resulting from MitoTracker Red staining) were observed in compartments distributed in the cytoplasm but not in the cell nuclei (figure 2a). The overlay of the figures 2a, 2b, and 2d is shown in 2c and exhibits a distribution of fluorescence signals as follows: a part of the lymphocytes indicates a green fluorescence in the cell membrane range and a mixed fluorescence in the cytoplasm. Merging the two fluorescence signals (green + red) results in a co-localizing orange fluorescence. This suggests a localization of the CAI-molecule in close vicinity to the mitochondria.
Confocal investigations of treated and untreated human peripheral lymphocytes. The green fluorescence signals (2b) originate from both the biotinylated-CAI-BioShuttle and intrinsic biotin after treatment with Streptavidin, Alexa Fluor® 488-solution. A strong red fluorescence signal of the mitochondrial staining by the used MitoTracker red is detectable. The overlay of the figures 2a and 2b as well as the corresponding DIC picture (2d) shows that the green fluorescence signal of the CAI-BioShuttle is co-localized with the red fluorescence of the mitochondrial compartment resulting in orange (mix fluorescence (2c). The bars indicate 20 µm.
At present, the reason for the mitochondrial colocalization is unknown. In order to support these results and to exclude a possible colocalization to lysosomes we also investigated the BioShuttle-CAI-peptide (figure 3) in HEL cells.
Confocal comparison of treated and untreated HEL cells. The green fluorescence signals originate from the biotinylated-CAI-BioShuttle treated cells (3a) after staining with Streptavidin, Alexa Fluor® 488-solution. The cells harbour an additional red fluorescence signal after lysosome-staining by LysoTracker red. The untreated HEL cells (3b) show the intrinsic biotin as low green fluorescence background. The bars indicate 20 µm.
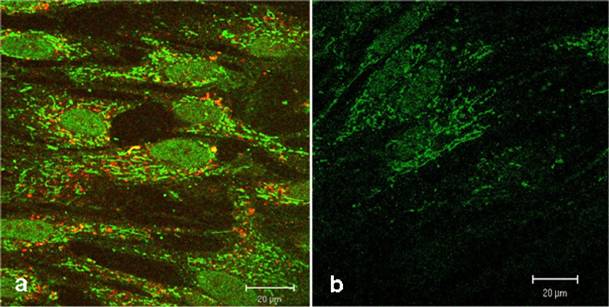
Figure 3a shows a strong green fluorescence signal derived from the CAI-peptide appearing to be lysosomally located, whereas the corresponding control (figure 3b) shows a very low diffuse signal originating from intrinsic biotin after treatment with Streptavidin, Alexa Fluor® 488-solution. The localization of active substances in lysosomes could alter the pharmacological property, which could lead to a loss of function by degradation with intra-lysosomal enzymes. To exclude this possibility, we used here the LysoTracker red staining. However figure 3a shows no significantly merged fluorescence signals, but instead distinct red lysosomes spatially separated from green fluorescence signals of the CAI-peptide in cytoplasm, mitochondria, and nuclei.
Conformational preferences of CAI-inhibitor and CAI-BioShuttle
In living cells the cytosolic reductive conditions are causing a cleavage of the coupling disulfide bridge of the CAI-BioShuttle and therefore the physico-chemical behavior of the free CAI-peptide is of particular interest. In order to gain some deeper insight into the conformational preferences of the free CAI-Inhibitor in solution we performed ultraviolet circular dichroism (CD) measurements to estimate important characteristics of its secondary structure. For comparison the secondary structure of both controls the REVCAI-Peptide (③) and the CAICTRL-Peptide (②) were determined also.
None of these peptides was soluble in distilled water at a concentration of 100µg/ml. The investigated peptides showed unequal solubility: whereas the control peptides could be dissolved as a 1 mg/ml stock solution in 10% TFE: 90% distilled water; the CAI-peptide (①) could only be dissolved in 100% TFE at 1 mg/ml; all probes were subsequently diluted to 100 µg/ml in 10% TFE. This water insoluble CAI-peptide was used for the CD measurements and revealed a strong β-sheet component (figure 4).
Polarity titration of the CAI-peptide. We performed a titration to measure the influence of TFE on the structure as described in methods. The relative amount of secondary structure motifs of the CAI-Inhibitor is monitored here by UV-CD polarity titration. The ordinate reveals the relative percentage of structure type. The abscissa shows the concentration of TFE in water.
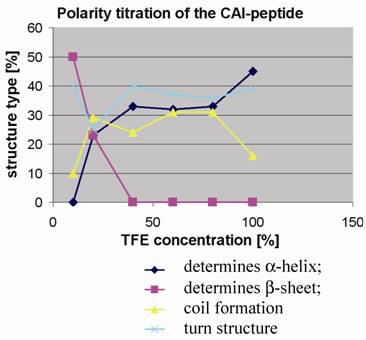
To determine to what extent the peptides were capable of adopting the expected α-helical conformation when environmental conditions were altered, as for example when fitting to a binding site, the peptides were titrated in trifluoroethanol (TFE): H2O mixtures and CD spectra were measured to monitor any changes in the relative structural content. TFE is an apolar solvent that is miscible with water and is known to stabilize intra-molecular hydrogen bonds in proteins and their fragments [20, 21].
At 10% TFE the CAI inhibitor (figure 4) shows about 50% regular β-strands, whereas the two control peptides contain high levels of coil and turn and a relatively low amount of regular secondary structure. The fact that the CAI peptide (figure 1;①) shows the poor solubility characteristics in water as described above strongly suggests that oligomeric aggregates are formed under these conditions, characteristic of β-structures in aqueous solution. The other three peptides (figure 1;②③④) behaved quite differently under polarity titration. The CAI-peptide (figure 1;①) is the only one capable of forming significant amounts of α-helical conformation and it is induced to do this at relatively low concentration of TFE. The three control peptides never formed large stretches of α-helical conformation and are not very sensitive to slight drops in polarity.
Coupling of the CAI-peptide to the BioShuttle transporter led to a much better solubility, the complex being soluble in pure water at 1 mg/ml. To investigate the influence of the BioShuttle-transporter-peptide coupled to the CAI on the conformational preferences of the CAI-peptide (figure 1;①) we performed UV CD measurements of the CAI-BioShuttle construct as well as on the inverse CAI-peptide (②) attached to the BioShuttle using the same experimental procedure described above.
Figure 5 shows for the CAI-BioShuttle peptide conjugate that the amount of regular structure is considerably reduced as compared to the free CAI-peptide (figure 1;①). More particularly, no β-strand is now present while there is a pronounced (~25%) α-helical component. The question arises whether this can be related to the CAI moiety only or whether the BioShuttle moiety can form a stable helix and therefore may contribute also to the α-helical component of the CD spectrum. An indication that the helical structure in the CAI-BioShuttle conjugate arises from the CAI is that the inverse CAI-peptide (figure 1;③) attached to the BioShuttle shows even less tendency to form regular secondary structure than the inverse CAI alone, the amount of α-helical conformation increasing linearly from zero at 10% TFE to only about 15% at 100 % TFE. The discovery that the free CAI peptide, although having a relatively short sequence, shows a pronounced tendency to adopt α-helical conformation under certain conditions coincides with other findings. It has been shown experimentally that the CAI-peptide exhibits a pronounced α-helical conformation for all 12 amino acids when binding to the HIV-1 capsid C-terminal domain (PDB ID: 2BUO). The bound conformation shows a high complementarity to the HIV surface.
Polarity titration of the CAI-BioShuttle. We performed a titration to measure the influence of TFE on the structure as described in methods. The relative amount of secondary structure motifs of the CAI-Inhibitor is monitored here by UV-CD polarity titration. The ordinate reveals the relative percentage of structure type. The abscissa shows the concentration of TFA in water.
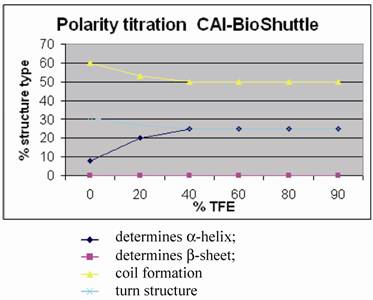
For the conformation of the BioShuttle transporter molecule alone an amphiphilic helix has been proposed [22, 23]. However no such helix is present in the TAT protein 3D structure solved by NMR where the BioShuttle peptide is a part of the sequence [24]. Our CD measurements also suggest that the BioShuttle moiety does not form a stable α-helix in solution.
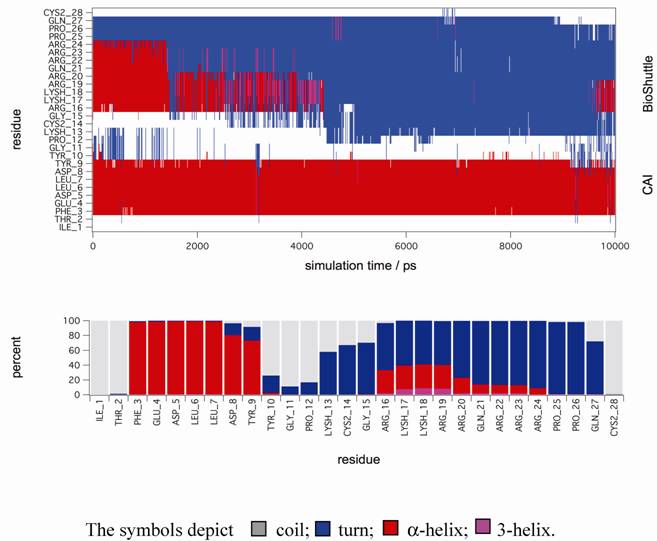
In order to gain further support for these assumptions MD simulations in explicit solvent were performed to gain deeper insight on the stability of secondary structure motifs of CAI-BioShuttle and CAI on the atomistic level. The starting structure of CAI-BioShuttle was built as an α-helix in order to check whether a helical structure for the BioShuttle moiety is stable in solution (see Material and Methods). For the free CAI peptide the helical conformation as present in the crystal structure was used as a starting structure. After about 2 ns simulation time the α-helix of the BioShuttle moiety starts to degrade (figure 6) whereas the helix of the CAI moiety remains completely stable over the whole simulation period of 10 ns (figure 7). The overall secondary structure statistics for the whole trajectory is about 30% α-helix, 45% turn and 25% coil and is in good agreement with the CD measurements. Three MD simulations of the free CAI-peptide in water were performed and the initial α-helix was stable for 10 ns (whole simulation period), 6 ns and 2 ns respectively (data not shown).
Molecular dynamic simulations. The figure shows a secondary structure analysis of a 10 ns MD simulation of CAI-BioShuttle. BioShuttle looses helical structure (shown in red) after about 2 ns simulation time and prefers a 'turn'–like (blue) orientation of the backbone torsions for the rest of the simulation time (top). Statistics of the secondary structure motifs per residue (bottom) showing that the probability for α-helical structure is low for the BioShuttle whereas the α-helical properties of the CAI moiety are very high.
Structural snapshot of the CAI-BioShuttle. The selected snapshot at the end of the 10 ns MD simulation shows the CAI adopting a conformation very close to the active conformation in the complex with the HIV-1 capsid C-terminal domain (PDB ID: 2BUO). The initial α-helix of the BioShuttle moiety disappeared.
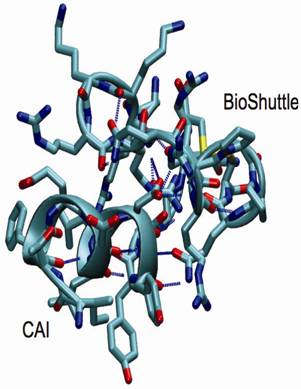
As a conclusion the MD simulations showed that CAI is able to exist in a α-helix in solution over a significant amount of time in contrast to the BioShuttle peptide for which the stability of the α-helix is significantly reduced. This is in excellent agreement with the CD measurements. However a simulation period of 10 ns is probably too short to draw definite conclusions on the conformational equilibrium. MD simulations covering a much longer (microsecond) time period are underway.
In silico interaction studies
A 'flexible docking' approach using AutoDock 3.05 [25] was applied to analyze whether the binding mode of the CAI to the HIV-1 capsid C-terminal domain (C-CA) receptor, as found in the crystal structure, could be reproduced in silico and whether there are alternative CAI conformations that could bind with a similar binding affinity. In a first approach we performed docking experiments where no pre-organization of the CAI-peptide was assumed, (so-called 'flexible docking' experiments where all torsion-angles of the peptide – except the peptide bonds - are allowed to adopt all possible conformations). Unfortunately it turned out that following the flexible docking approach we were not able to find any conformation, which was bound in a similar conformation or was bound as tightly as the one reported in the X-ray structure. The number of docking experiments performed using a genetic search algorithm can be regarded as very high (see Material and Methods) and are clearly at the limit on what is technically feasible at the moment. The reason why the conformation of CAI as present in the X-ray structure could not be reproduced even in a extensive flexible docking experiment is clearly because the bound conformation is highly organized and has therefore, because of the many rotatable bonds, a very low probability of being found in an unbiased search. Only when the conformation of the core peptide backbone was pre-organized as an α-helix, complexes very similar to the crystal structure could be obtained from (semi-flexible) docking experiments. If was found that the binding energy of CAI was more favorable for the helical conformations than for the more hairpin-like conformations which were mainly adopted in 'best poses' of the unbiased search.
Evaluation of the individual energy contributions to the binding free energy as derived from the AutoDock scoring function revealed a very unfavorable torsional term for the binding process. Because of the many bonds which can freely rotate in the free peptide, the loss in entropy when freezing out the rotations upon binding to the protein surface can evidently not be compensated by the gain of enthalpy on binding, so that the scoring function of the docking program used indicated no or only weak binding.
These findings suggest that the binding affinity would dramatically benefit if the free CAI-peptide would have an intrinsic tendency to form an α-helical conformation, so that in the conformational ensemble present in solution, a significant amount of molecules would be pre-organized for binding already. In such a way, the loss of entropy in freezing out the specific conformation required for binding would be minimal.
Summary
The transfer of CAI-peptides across biomembranes is very poor and needs transporter molecules which can separate the CAI-cargo after the transmembrane passage in order to exclude undesired side effects like sterical interactions with the CAI-peptide cargo at the target site. The coupling of peptides to a BioShuttle carrier increased the bioavailability inside the cell. Intensive docking studies did not reveal alternative CAI conformations that are strongly interacting and failed to reproduce the binding mode of the crystal structure when the backbone of the CAI-peptide was not pre-organized as an α-helix. The reason for this is the high number of rotatable bonds in the peptide and the high specificity of the CAI-receptor interaction that can only be satisfied when the peptide is properly folded. However a complex very similar to the crystal structure could be reproduced by docking experiments when the CAI backbone was pre-organized as an α-helix. Therefore it can be assumed that only one highly specific conformation for strong binding to C-CA exists. The CD measurements and MD simulations suggest that an intrinsic α-helical conformation of the isolated CAI-peptide may exist, and is obviously not significantly hampered by attaching the BioShuttle peptide to enable transport through the membrane. Such a preferred folding of the CAI inhibitor seems to be an important factor for high affinity binding, since the entropic penalty for forming the required conformation on binding to CA is considerably reduced. CD measurements revealed that the reverse and scrambled peptide do not show such a pre-organization which can explain their inactivity. Further studies with CAI-BioShuttle transporter should be considered for additional or alternative antiretroviral interventions.
Material and Methods
Chemical Synthesis and Purification of the CAI-BioShuttle
For solid phase synthesis of the modules of CAI-BioShuttle and the control probes (figure 1) we employed the Fmoc-strategy [26, 27] in a fully automated multiple synthesizer (Syro II, MultiSyntech). Peptide chain assembly was performed using in situ activation of amino acid building blocks by 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU). The biotin was built-in on the ε-amino group of lysine.
The intermediates and products were purified by preparative HPLC on an YMC-Pack ODS. 5µm 120A reverse phase column (20 × 150 mm) using an eluent of 0,1% trifluoroacetic acid in water (A) and 80% acetonitrile in water (B). The peptides were eluted with a successive linear gradient of 25% B to 80% B in 30 min at a flow rate of 10 ml/min. The fractions corresponding to the purified proteins were lyophilized. The purified material was characterized with analytical HPLC and laser desorption mass spectrometry (purity >90%) Reflex II (Bruker).
The four different 14-mer peptide-modules, shown in figure 1, were oxidized together with the transmembrane transport module in the range of 2 mg × ml-1 in a 20% DMSO water solution. The reaction was completed after 5 hours. The formation of the sulfur bridge was controlled with matrix assisted laser desorption mass spectrometry Reflex II (Bruker). The mass spectra of the investigated CAI-BioShuttle-constructs are represented in figure 1.
Cell culture
We obtained the human peripheral lymphocytes (PTL) from Institute of Pathology, University of Heidelberg. PTL were isolated from 10 ml native venous blood from a healthy donor by a lymphocyte preparation with Lymphoprep™ gradient (AXIS-Shield PoC AS, Oslo Norway) under sterile conditions maintained in RPMI 1640 supplemented with G-CSF and human embryonic lung cells (HEL) (obtained from DKFZ Tumorbank) in RPMI 1640 Medium without phenol red complemented with 10 % fetal bovine serum (FBS), (Gibco BRL). The cell cultures were grown at 37°C and 5 % CO2.
Cell preparation for confocal laser scanning microscopy (CLSM)
Lymphocytes
For estimation of the intracellular localization of the CAI-BioShuttle, four cell culture flasks with lymphocytes and 2 ml RPMI/G-CSF medium were incubated in parallel with the CAI-BioShuttle constructs (figure 1;①) 1 h in a 100 nM final concentration. After the cells were washed and resuspended in phenol red free RPMI medium, the cell suspensions in a volume of 100 µl were added to the glass slides (Lab Tek® II; Chamber Slide™ System). Their glass surface was treated with the BD Cell-Tak™ Cell and Tissue Adhesive (BD Biosciences) before immobilization of the suspension cells according to the instructions. The immobilized living lymphocytes were stained with Mito Tracker Red (Molecular Probes) for 1 hour and the cell containing slide sections were rinsed twofold gently with Hanks (Gibco) before and after the fixation procedure with 3.7 % paraformaldehyde (PFA) for 15 minutes at room temperature. The cell membranes were slightly perturbed by treatment with Triton X-100 solution (0.1 % in Hank's) on ice for 2 minutes, followed by twice washing the cells. Then 150 µl of Streptavidin, Alexa Fluor® 488-solution (1:100 in PBS) (Molecular Probes) were applied to cells over 45 minutes at room temperature. The unbound Streptavidin-conjugate was removed with Hank's solution, again rinsed twice with Hank's and embedded in Vectashield®Mounting Medium (Vector Laboratories). The intracellular distribution of the Biotin-labeled- Streptavidin Alexa Fluor® 488 CAI-BioShuttle constructs was verified using a Zeiss Laser confocal microscope (LSM 510 UV). The optical slice thickness was 700 nm.
HEL cells
Adherent HEL cells were grown as described above and fixed as shown for the lymphocytes.
The excitation line of an Argon laser was used to detect the fluorescence signal from the Biotin-labeled/Streptavidin Alexa Fluor® 488-labeled CAI-BioShuttle-conjugate. The spatial organization of lysosomes was shown by use of the LysoTracker Red fluorescence with the Zeiss Filter Set 31 (578 nm excitation and 599 nm emission). To increase the contrast of the optical sections, 12–20 single exposures were averaged. The image acquisition parameters were adapted to show signal intensities in accordance with the visible microscopic image. The same experiments were performed with scrambled random sequence-constructs and their reverse amino acid sequence as controls respectively (figure 1).
Circular Dichroism Studies
Far UV circular dichroism spectra were measured from 190-240 nm using a Jasco J-710 automatic recording spectral polarimeter calibrated with 0.05% β-androsterone in dioxane. The scanning speed was 5.0 nm/minute with a 4.0 s time constant. Spectra displayed result from four-fold signal averaging followed by Fast Fourier Transform to remove residual noise; similarly treated baselines were subtracted before converting from millidegrees to θmrw (mean residue ellipticity) for secondary structure analysis using the computer program PEPFIT [28]. Samples were measured in a 1.0 mm dichroically neutral quartz cuvette at a concentration of 100 µg/ml. TFE titration ran from 0% to 100% TFE in 20% steps with distilled water as the aqueous component.
Molecular Dynamics simulation
3D coordinates of the HIV-1 capsid C-terminal domain (C-CA) in complex with CAI [16] were retrieved from the Protein Data Bank [29] (PDB ID: 2BUO, resolution 1.7 Å). The coordinates of the CAI peptide were extracted and were used as starting conformation for MD simulations of the free peptide in water. The BioShuttle peptide (GRKKRRQRRRPPQC) and the elongated CAI sequence (ITFEDLLDYYGPKC) were built separately from AMBER building blocks using the LEAP module of the AMBER package [30]. Linking of the two chains by forming a disulfide bond between the C-terminal cysteines and folding of the molecule into the starting conformation was performed using the Conformational Analysis Tools (CAT) [31] software applying the method briefly outlined here: for the CAI fragment the torsion angles were extracted from the crystal structure and imposed on the ITFEDLLDYYGP sequence, the rest of the chain was folded into an α-helix by setting the φ/ψ torsions to -57°/-47° respectively. The peptides were solvated in a box of SPC water and ions were added to counterbalance the charge of the peptides. The particle-mesh Ewald approach was used to account for long-range electrostatic effects. Temperature and pressure was held constant at 300 K and 1 bar using Berendsen methods [32]. All MD simulations were performed for 10 ns using the GROMACS package and the GROMOS96 forcefield [33]. Analysis of the stability of secondary structure motifs during the MD simulation has been performed using STRIDE [34] interfaced with CAT. Igor Pro (www.wavemetrics.com) has been used to generate the scientific plots. VMD [35] was used for molecular graphics.
Flexible Docking
AutoDock 3.05 was used to perform the docking experiments [25]. The various files required as input for AutoDock were created with the help of 'AutoDockTools' (http://www.scripps.edu/~sanner/python/adt/). The genetic algorithm with local search option (GA-LS) as implemented in AutoDock was used to dock the flexible peptide. For the 'flexible' docking experiments backbone (φ/ψ) and side chain torsions were allowed to rotate (in total 32 torsions which is the maximum number of flexible torsions that AutoDock can handle in the standard installation) whereas in the 'semi flexible' docking experiments only the side chain torsions were allowed to rotate. The receptor was treaded as rigid for the docking experiments. For the 'flexible' docking 333 AutoDock jobs were started on a HPC cluster (AMD Opteron 250 processor with 2.4 MHz) each performing 256 GA-LS runs (106 energy evaluations each) giving rise to 85248 docked CAI structures. The overall CPU time was about 7000 hours. The docking protocol with semi-flexible CAI implied 44 AutoDock jobs resulting in 11264 docked solutions. CAT was used to merge the output data of the AutoDock runs, to perform the analysis of the entire dataset and organize the results in such a way, that complexes exhibiting a strong binding can be easily visualized using standard display programs.
Acknowledgements
The authors wish to thank Gabriele Müller, Ulrike Bauder-Wuest and Andrea Breuer for excellent technical assistance with the CAI-studies. Additionally we thank Christine Otto and Jochen vom Brocke for continuous support and critical discussions for improving the language of the manuscript.
Abbreviations
CA: Capsid; CAI: Capsid Asembly Inhibitor; CAT: Conformational Analysis Tools; CD: Circular Dicroism; CLSM: Confocal Laser Scanning Microscopy; HAART: Highly Active Antiretroviral Therapy; HEL: Human Embryonic Lung Cell Line; HIV: Human Immunodeficiency Virus; MA: Matrix; MD: Molecular Dynamics; NC: Nucleocapsid; PTL: Peripheral T Lymphocytes.
Conflict of Interest
We declare no conflicts of interest.
References
1. UNAIDS. Report on the global AIDS epidemic. 2007 AIDS epidemic update. UNAIDS publications. 2008;1:3-43
2. Richman DD. HIV chemotherapy. Nature. 2001;410:995-1001
3. Vierling P, Greiner J. Prodrugs of HIV protease inhibitors. Curr Pharm Design. 2003;9:1755-70
4. Rathbun RC, Lockhart SM, Stephens JR. Current HIV treatment guidelines--an overview. Curr Pharm Des. 2006;12:1045-63
5. Tamalet C, Yahi N, Tourres C. et al. Multidrug resistance genotypes (insertions in the beta3-beta4 finger subdomain and MDR mutations) of HIV-1 reverse transcriptase from extensively treated patients: incidence and association with other resistance mutations. Virology. 2000;270:310-6
6. Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1-15
7. Bryant M, Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A. 1990;87:523-7
8. Navia MA, Fitzgerald PM, McKeever BM. et al. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature. 1989;337:615-20
9. Ganser-Pornillos BK, Yeager M, Sundquist WI. The structural biology of HIV assembly. Curr Opin Struct Biol. 2008;18:203-17
10. Wiegers K, Rutter G, Kottler H. et al. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J Virol. 1998;72:2846-54
11. Tang C, Loeliger E, Kinde I. et al. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol. 2003;327:1013-20
12. Kelly BN, Kyere S, Kinde I. et al. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J Mol Biol. 2007;373:355-66
13. Li F, Zoumplis D, Matallana C. et al. Determinants of activity of the HIV-1 maturation inhibitor PA-457. Virology. 2006;356:217-24
14. Martin DE, Blum R, Doto J. et al. Multiple-dose pharmacokinetics and safety of bevirimat, a novel inhibitor of HIV maturation, in healthy volunteers. Clin Pharmacokinet. 2007;46:589-98
15. Sticht J, Humbert M, Findlow S. et al. A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol. 2005;12(8):671-7
16. Ternois F, Sticht J, Duquerroy S. et al. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat Struct Mol Biol. 2005;12:678-82
17. De Clercq E. New anti-HIV agents and targets. Med Res Rev. 2002;22:531-65
18. Lee RJ, Huang L. Lipidic vector systems for gene transfer. Crit Rev Ther Drug Carrier Syst. 1997;14:173-206
19. Braun K, Peschke P, Pipkorn R. et al. A biological transporter for the delivery of peptide nucleic acids (PNAs) to the nuclear compartment of living cells. J Mol Biol. 2002;318:237-43
20. Graf von SA, Jimenez MA, Kinzel V. et al. Solvent polarity-dependent structural refolding: a CD and NMR study of a 15 residue peptide. Proteins. 1995;23:196-203
21. Shiraki K, Nishikawa K, Goto Y. Trifluoroethanol-induced stabilization of the alpha-helical structure of beta-lactoglobulin: implication for non-hierarchical protein folding. J Mol Biol. 1995;245:180-94
22. Joliot A, Prochiantz A. Transduction peptides: from technology to physiology. Nature Cell Biology. 2004;6:189-96
23. Loret EP, Vives E, Ho PS. et al. Activating region of HIV-1 Tat protein: vacuum UV circular dichroism and energy minimization. Biochemistry. 1991;30:6013-23
24. Peloponese JM Jr, Gregoire C, Opi S. et al. 1H-13C nuclear magnetic resonance assignment and structural characterization of HIV-1 Tat protein. C R Acad Sci III. 2000;323:883-94
25. Morris GM, Goodsell DS, Halliday RS. et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19:1639-62
26. Merriefield RB. Solid Phase Peptide Synthesis. I The Synthesis of a Tetrapeptide. J Americ Chem Soc. 1963;85:2149-54
27. Paquet A. Introduction of 9-fluorenylmethoxycarbonyl, trichloroethoxycarbonyl, and benzyloxycarbonyl amine protecting groups into O-unprotected hydroxyamino acids using succinimidyl carbonates. Can J Chem. 1982;60:976-80
28. Reed J, Reed TA. A set of constructed type spectra for the practical estimation of peptide secondary structure from circular dichroism. Anal Biochem. 1997;254:36-40
29. Berman HM, Westbrook J, Feng Z. et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235-42
30. Case DA, Cheatham TE III, Darden T. et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668-88
31. Conformational Analysis Tools (CAT). : Frank M. http://www.md-simulations.de/CAT/
32. Berendsen HJC, Postma JPM, van Gunsteren WF. et al. Molecular Dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684-90
33. van der SD, Lindahl E, Hess B. et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701-18
34. Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566-79
35. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33-8
Author contact
![]() Correspondence to: Dr. Klaus Braun, German Cancer Research Center (DKFZ), Dept. of Molecular Toxicology, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany. Phone: ++49 6221-42 2495; Fax: ++49 6221-42 2442; e-mail: k.braunde
Correspondence to: Dr. Klaus Braun, German Cancer Research Center (DKFZ), Dept. of Molecular Toxicology, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany. Phone: ++49 6221-42 2495; Fax: ++49 6221-42 2442; e-mail: k.braunde