Impact Factor ISSN: 1449-1907
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Background
2. Methods
3. Results and Discussion
4. Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2007; 4(4):232-236. doi:10.7150/ijms.4.232 This issue Cite
Short Research Communication
Familial Polycythemia Caused by a Novel Mutation in the Beta Globin Gene: Essential Role of P50 in Evaluation of Familial Polycythemia
Neeraj Agarwal 1, Mariluz P. Mojica-Henshaw 2, Elizabeth. D. Simmons 3, Dottie Hussey 4, Ching N. Ou 5, Josef T. Prchal 6 ![]()
1. Department of Internal Medicine, University of Utah, Salt Lake City, Utah, USA
2. Department of Internal Medicine, University of Utah, Salt Lake City, Utah, USA
3. Department of Hematology and Oncology, Kaiser Permanente, Southern California, USA
4. ARUP Laboratories, Salt Lake City, Utah, USA
5. Department of Pathology, Texas Children's Hospital. Houston, Texas, USA
6. Department of Internal Medicine, University of Utah, Salt Lake City, Utah & ARUP Laboratories, Salt Lake City, Utah, USA
Received 2007-8-3; Accepted 2007-10-3; Published 2007-10-4
Abstract
Two polycythemic subjects from a family with multiple polycythemic subjects were evaluated. Estimation of oxygen affinity of Hb from venous blood gas parameters (P50) revealed low P50 suggesting a high affinity Hb variant. Further work up, which included beta globin gene sequencing, revealed a novel mutation changing a codon to the previously reported high affinity Hb - Hb Johnstown (beta109 Val->Leu). Polycythemic subjects with high affinity Hb variant are asymptomatic with normal life expectancy. Their differentiation from polycythemia vera (PV) is crucial to avoid therapy which is otherwise reserved for PV patients. We provide an electronic version (in Microsoft excel program) of a previously reported mathematical formula for rapid calculation of P50 from venous blood gases. Estimation of P50 is an essential initial step in the evaluation of a subject with personal and family history of polycythemia.
Keywords: High affinity Hb, Polycythemia, Estimation of P50
1. Background
Polycythemia means many cells in the blood. Polycythemia is clinically defined as elevated red blood cell mass ( hemoglobin > 18.5 g/dL in men, 16.5 g/dL in women or other evidence of increased red cell volume or hemoglobin or hematocrit greater than 99th percentile of method-specific reference range for age, sex, altitude of residence or hemoglobin greater than 17 g/dL in men, 15 g/dL in women if associated with a documented and sustained increase of at least 2 g/dL from an individual's baseline value that can not be attributed to correction of iron deficiency, or elevated red cell mass greater than 25% above mean normal predicted value [1].
Polycythemia can be further classified as primary polycythemia, secondary polycythemia, or polycythemia due to abnormal hypoxia sensing.
Primary polycythemias are caused by intrinsic defects in the erythroid precursors that result in hyper responsiveness to normal level of serum erythropoietin (Epo). Secondary polycythemias are driven by the factors (predominantly Epo but also insulin growth factor 1 and cobalt) extrinsic to the erythroid progenitor cells. Generally, in secondary polycythemia, the increased red cell mass represents a physiologic response to tissue hypoxia or abnormally increased level of serum Epo [2].
Polycythemias due to abnormal hypoxia sensing include Chuvash polycythemia, polycythemias associated with von Hippel-Lindau mutations other than the Chuvash polycythemia mutation, and polycythemia due to proline hydroxylase mutation [3, 4]. Acquired conditions that lead to increased Epo production, such as chronic hypoxia and a variety of tumors, are the most common causes of secondary polycythemias.
Secondary congenital polycythemia results from inherited conditions that lead to increased Epo levels. These include hemoglobin variants with high affinity for oxygen, congenitally low erythrocyte 2, 3 biphosphoglycerate levels, and inherited methemoglobinemias. All these conditions are characterized by a left shift in Hb dissociation curve which in turn leads to tissue hypoxia and a physiologically appropriate increase in Epo levels. Congenital cyanotic heart or lung disorders, which lead to tissue hypoxia and increased Epo level are also examples of secondary congenital polycythemias but these are characterized by a normal Hb dissociation curve.
2. Methods
Two subjects (mother and daughter), from a family with multiple subjects with polycythemia, were available for evaluation:
Subject 1(mother): She is a 59 year old Caucasian woman who presented with lifelong history of polycythemia. Her medical history was remarkable for 1) multifocal ductal carcinoma in situ (DCIS) of the breast in 2002, treated with simple mastectomy and 2) colonic diverticula and internal hemorrhoids since 2005. Family history was remarkable for presence of lifelong history of polycythemia in her mother and two daughters. Physical examination was normal (no hepatosplenomegaly). Laboratory parameters revealed high Hb (16 gm%) , high hematocrit (48%), normal MCV, normal platelet and white blood cell counts, normal arterial oxygen saturation, and normal liver function tests.
Subject 2 (daughter): She is a 30 year old Caucasian woman who presented for further evaluation of lifelong history of polycythemia. She had mild fatigue but no other symptoms. Past medical history was unremarkable. Family history was significant for lifelong history of polycythemia in her mother, maternal grandmother and her younger sister. Physical examination was unremarkable. Laboratory parameters were remarkable for elevated hemoglobin (17.2 gm%) and hematocrit (51.4%) but normal MCV, normal platelets and white blood cell counts, normal arterial oxygen saturation, and normal liver function tests.
Both subjects underwent following laboratory tests: Serum Epo levels were normal in both subjects (15 mIU/ml and 19 mIU/ml respectively, normal range 4.1 - 19.5 mIU/ml). Venous blood gas parameters were obtained which included partial pressure of oxygen (venous pO2), venous pH and venous oxygen saturation. Calculation of affinity of Hb for oxygen (P50) was done using the mathematical formula as described [5]. Routine Hb electrophoreses, high performance liquid chromatography (HPLC) [6, 7], isoelectric focusing (IEF) in polyacrylamide gel, globin chain analysis [8] and peptide mapping [9] were performed. This was followed by beta globin gene sequencing which was performed by ABI3730 96-capillary sequencer at the DNA Sequencing Core Facility at the University of Utah School of Medicine (primers and conditions available upon request).
3. Results and Discussion
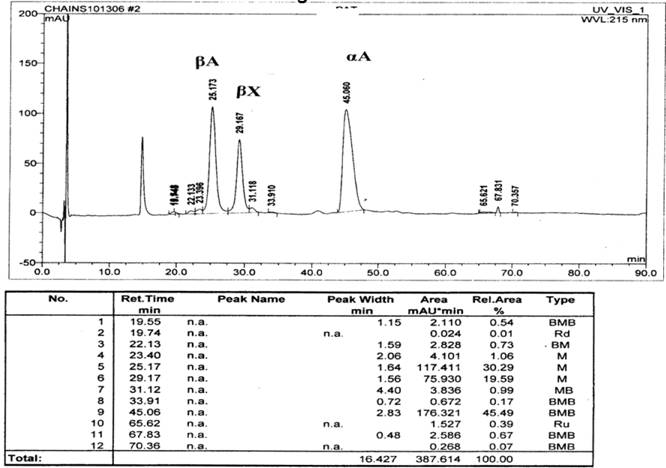
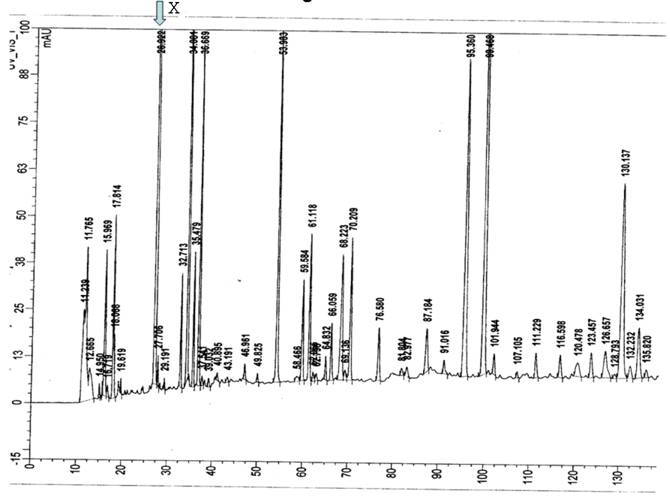
The P50 was found to be low at 18 mm Hg (normal range 22.6 to 29.4) suggesting increased affinity of Hb for oxygen. Routine Hb electrophoresis and HPLC failed to detect mutant hemoglobin. IEF showed a band anodal to Hb A and globin chain analysis by HPLC revealed an unidentified beta globin variant in both subjects (Figure 1). Peptide mapping showed an extra peak at 26.9 min but showed no decrease in any peaks suggesting a mutation somewhere in the core (Figure 2). Beta globin gene sequencing revealed a novel mutation (GTG->TTG ) of codon 109 of exon 3 of beta globin gene. This mutation leads to a previously reported high affinity Hb variant known as Hb Johnstown (beta109 Val->Leu) [10-12]; however, this nucleotide change is novel and previously unreported; and it leads to a previously described amino acid substitution that was however, caused by a different nucleotide missense mutation.
HPLC globin chain separation. Y axis denotes relative mass, X axis denotes the retention time in minutes. The unidentified beta globin variant is shown as beta X.
HPLC peptide mapping. Y axis denotes relative mass, X axis denotes the retention time in minutes. An extra peak at 26.9 min (X) was seen without decrease in any other peaks suggesting a mutation somewhere in the core.
Hb Johnstown (beta109 (G11) Val->Leu) is a high oxygen affinity Hb variant and there are three reports in the literature. It was first reported by Jones and colleagues in Oregon, in 1990, in a healthy asymptomatic subject with mild erythrocytosis and left-shifted hemoglobin-O2 dissociation curve[10]. As with many other Hb variants, Hb Johnstown is silent on standard hemoglobin electrophoretic analyses, and was identified and isolated by reverse-phase HPLC of individual globin chains. Structural analysis revealed the substitution beta 109 (G11) Val ->Leu [10]. In 2000, the underlying beta globin mutation [beta-globin codon 109 (GTG ->CTG )] was first reported by Ropero and colleagues, in two unrelated families (total four subjects) of Basque extraction in Spain [11]. In one of these families, Hb Johnstown mutation was present in double heterozygosity with another beta 0 thalassemia mutation IVS-1-nt1 (G->A). In 2004, Feliu-Torres and colleagues reported Hb Johnstown [beta-globin codon 109 (GTG ->CTG )] in an eight year old girl, who had been referred for evaluation of erythrocytosis, in double heterozygosity with another beta globin mutation [IVS-I-1(G->A)]. Her asymptomatic mother was found to be heterozygous for Hb Johnstown mutation [12]. However, these reports described a causative G to C mutation that is different that mutation we describe in our subject with Hb Johnstown, namely G to T, both encoding the same amino acid; i.e. leucine and present in subjects not known to be of Spanish/Basque extraction.
Oxygenation and deoxygenation of hemoglobin occur at the heme iron. The sigmoid shape of Hb-oxygen dissociation curve is indicative of cooperative interaction between heme and oxygen. Oxygen affinity and Hb-oxygen dissociation is affected by blood pH, 2, 3- biphosphoglycerate (2, 3 BPG) level in the red cell and temperature, and globin structure [13].
Affinity of Hb with oxygen is expressed as the P50, which is the partial pressure of oxygen in blood at which 50% of the Hb is saturated with oxygen. The venous P50 can be measured directly using a cooximeter which is no longer easily available in routine and even reference laboratories. Lichtman and colleagues have reported a mathematical formula which can be used to calculate P50 reliably [5]. Calculating P50 using this formula requires the following venous gas parameters: partial pressure of oxygen (venous pO2), venous pH and venous oxygen saturation, and uses anti-log mathematical function that many clinicians find difficult to use for calculation [5]. The P50 of a healthy person with normal Hb is 26 ± 1.3 mm Hg. The 99% confidence interval for individual observations has been reported to be 22.6 to 29.4 mm Hg. An abnormally low P50 reflects an increased affinity of hemoglobin for oxygen and vice versa. Elevations and reductions in 2, 3- BPG level in the erythrocyte will also lead to corresponding changes in P50 values; however, in only reported subjects this decrease was limited to a P50 value between 20 and 35 mm Hg. There should be high suspicion for the presence of a high affinity Hb variant if P50 value is <20 mm Hg [5].
During oxygenation and deoxygenation, there is considerable movement along the interface of alpha 1 and beta 2 chains of the Hb tetramer. Several hemoglobin variants have substitutions affecting this interface. All these substitutions can affect the cooperative nature of oxygen binding with heme, and in turn, can change the affinity of Hb for oxygen. The majority of mutations affecting oxygen affinity result in high affinity Hb variants which result in leftward shift of the dissociation curve and relative tissue hypoxia [14]. There are 90 high affinity Hb variants, listed on the globin server, known to be associated with high affinity for oxygen (http://globin.bx.psu.edu/hbvar/menu.html accessed on May 04, 2007) [15]. All these Hb variants are inherited in an autosomal dominant manner. High affinity Hb variants release oxygen in the tissue relatively slowly and create relative tissue hypoxia. This leads to increased production of Epo from kidneys which results in increased red blood cell mass and polycythemia. At an elevated level of increased red blood cell mass (which depends upon the oxygen affinity of a given variant) adequate oxygenation of the tissue is reestablished and Epo production plateaus and at this new steady state serum Epo is often at normal level. This leads to stabilization of Hb level after achieving a certain elevated level of hematocrit.
4. Conclusion
Family history of polycythemia in a subject with polycythemia should raise the suspicion for the presence of a high affinity Hb variant [14]. A low P50 value (obtained from venous gas parameters) is supportive of high oxygen affinity Hb variant or decreased 2, 3 BPG level. In a polycythemic patient, establishing a correct diagnosis of a high affinity Hb variant is important as these patients have normal life expectancy and do not require phlebotomy. The therapies used for polycythemia vera such as phlebotomy and chemotherapy should not be used in patients who have polycythemia due to high affinity Hb variants. With this report, we are providing an electronic version (using Microsoft Excel program) of the mathematical formula [5], with which P50 can be calculated in few seconds, provided venous gas parameters are available, without necessity of more sophisticated calculations using antilog parameters. With increased ease and rapidity of calculation using our Excel program (Supplementary Material), we hope that use of P50 will increase, leading to improved detection of Hb variants in subjects with familial polycythemia.
Supplementary Material
Excel program
Acknowledgements
Ken J. Bulpitt, M.D, developed the electronic program in Microsoft Excel program (Supplementary Material) from the previous reported mathematical formula for the calculation of P50. This work was supported in part by National Institutes of Health research Grants R01HL5007-09 (J.T.P.).
Conflict of interest
Financial disclosure: Consultant: Josef T. Prchal for Astra Zeneca, Amgen; Honoraria: Josef T. Prchal for Astra Zeneca, Amgen. The authors declared that no other conflict of interest exists.
References
1. Tefferi A, Thiele J, Orazi A. et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110:1092-1097
2. Prchal J. Primary polycythemias. Curr Opinion Hematol. 1995;2:146-152
3. Gordeuk V, Stockton DW, Prchal JT. Congenital polycythemias/ erythrocytoses. Haematologica. 2005;90:109-116
4. Ang S, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, Sergueeva AI, Miasnikova GY, Mole D, Maxwell PH, Stockton DW, Semenza GL, Prchal JT. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32:614-621
5. Lichtman M, Murphy MS, Adamson JW. Detection of mutant hemoglobins with altered affinity for oxygen. A simplified technique. Ann Intern Med. 1976;84:517-520
6. Ou C, Buffone GJ, Reimer GL, Alpert AJ. High-performance liquid chromatography of human hemoglobins on a new cation exchanger. J Chromatogr. 1983;266:197-205
7. Ou C, Rognerud CL. Rapid analysis of Hb variants using a cation-exchange HPLC method. Clin Chem. 1993;39:820-824
8. Shelton JB, Shelton JR, Schroeder WA. High performance liquid chromatographic separation of globin chains on a large-pore C4 column. J Liquid Chromatogr. 1984;7:1969-77
9. Schroeder W, Shelton JB, Shelton JR, Powars D. Separation of peptides by high-pressure liquid chromatography for the identification of a hemoglobin variant. J Chromatogr. 1979;174:385-392
10. Jones R, Saiontz HI, Head C, Shih DT, Fairbanks VF. Hb Johnstown [beta 109 (G11) Val----Leu]: a new electrophoretically silent variant that causes erythrocytosis.1. Hemoglobin. 1990;14(2):147-156
11. Ropero P, Villegas A, Gonzalez AF, Anguita E, Sanchez J, Carreno DL, Arrizabalaga B, Atuxta L. Hb Johnstown [beta 109 (G11) Val-->Leu]: second case described and associated for the first time with beta(0)-thalassemia in two Spanish families. Am J Hematol. 2000;65(4):298-301
12. Feliu-Torres A, Eberle SE, Roldan A, Gonzalez S, Sciuccati G. Hb Johnstown [beta109(G11)Val-->Leu]: A high oxygen affinity variant associated with beta0-thalassemia. Hemoglobin. 2004;28(4):335-338
13. Bunn H, Forget BG. Hemoglobin: Molecular, Genetic and Clinical Aspects. Philadelphia: WB Saunders. 1986
14. Wajcman H, Galacteros F. Hemoglobins with high oxygen affinity leading to erythrocytosis. New variants and new concepts. Hemoglobin. 2005;29:91-106
15. Giardine B, van Baal S, Kaimakis P, Riemer C, Miller W, Samara M, Kollia P, Anagnou NP, Chui DH, Wajcman H, Hardison RC, Patrinos GP. HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum Mutat. 2007;28:206
Author contact
![]() Correspondence to: Josef T. Prchal, MD. 5C 310, SOM, 50 North Medical Drive, Salt Lake City, Utah, 84132. Josef.prchalutah.edu
Correspondence to: Josef T. Prchal, MD. 5C 310, SOM, 50 North Medical Drive, Salt Lake City, Utah, 84132. Josef.prchalutah.edu