Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2007; 4(4):223-231. doi:10.7150/ijms.4.223 This issue Cite
Research Paper
Chlamydia trachomatis Infection of Human Trophoblast Alters Estrogen and Progesterone Biosynthesis: an insight into role of infection in pregnancy sequelae
Anthony A. Azenabor, Patrick Kennedy, Salvatore Balistreri ![]()
Department of Health Sciences, University of Wisconsin, Milwaukee, WI 53211, USA
Received 2007-6-27; Accepted 2007-9-5; Published 2007-9-6
Abstract
The trophoblast cells are in direct contact with endometrial tissues throughout gestation, playing important early roles in implantation and placentation. The physiologic significance and the operating mechanisms involved in probable altered trophoblast functions following Chlamydia trachomatis infection were investigated to determine if C. trachomatis initiates productive infection in trophoblast, effects of such event on the biosynthesis of cholesterol and its derivatives estrogen and progesterone; and the regulator of the biosynthesis of these hormones, human chorionic gonadotropin. Chlamydia trachomatis exhibited productive infection in trophoblast typified by inclusion formation observed when chlamydia elementary bodies were harvested from trophoblast and titrated onto HEp-2 cells. Assessment of the status of C. trachomatis in trophoblast showed a relative increase in protein of HSP-60 compared with MOMP, features suggestive of chlamydial chronicity. There was a decrease in cellular cholesterol of chlamydia infected trophoblast and a down regulation of HMG-CoA reductase. The levels of estrogen and progesterone were decreased, while the expression of aromatase and adrenodoxin reductase was up regulated. Also, there was a decrease in human chorionic gonadotropin expression. The implications of these findings are that C. trachomatis infection of trophoblast may compromise cellular cholesterol biosynthesis, thus depleting the substrate pool for estrogen and progesterone synthesis. This defect may impair trophoblast functions of implantation and placentation, and consequently affect pregnancy sequelae.
Keywords: Chlamydia and pregnancy outcome, Chronic chlamydia in trophoblast, Steroid hormones, Trophoblast function
1. Introduction
Trophoblast, the first cell to differentiate from the fertilized egg is an invasive, eroding and metastasizing cell that exhibits a crucial role in implantation and placentation [1, 2, 3]. Preceding the invasion event, the uterine mucosa is transformed in a process called decidualization. It is such suitable environment that allows the differentiation of trophoblast in the villous and extravillous pathways [4]. The fulfillment of the enormous role of ensuring proper implantation and placentation requires a number of functional characteristics inherent in the trophoblast. These functions include; neural and endothelial functions [5, 6], phospholipids signaling function [7], endocrine functions [8], and immunocyte function. It stands to reason therefore that trophoblast injury will mediate impairment of these functions and degenerate into disturbed implantation and placentation [9]. Therefore, there is a compelling need to understand the role of a potential trophoblast injury mediator, such as a prevailing chlamydial infection afflicting the reproductive system. It is such need that this research addresses.
Chlamydia trachomatis is of significant importance as a cause of human diseases including, trachoma [10], infertility [11], salpingitis and ectopic pregnancy [12]. Chlamydiae are strict intracellular pathogens that exert enormous metabolic pressure on cells. Their life cycle is biphasic; the extracellular infectious form is the elementary bodies (EBs), which are metabolically inert. When they infect susceptible host cells, they transform into the reticulate bodies (RBs), which are the vegetative form of the organism, capable of metabolic activities and replicate intracellularly [13]. Pathophysiologic changes resulting from C. trachomatis affliction of cells are well documented, and importantly, C. trachomatis is the most prevalent bacterial cause of sexually transmitted diseases [14, 15]. Evidences abound that chlamydiae infection may cause human abortion by unknown mechanism [16, 17].
In this study, we reasoned that chronic C. trachomatis affliction of the female genitalia which ascends into the uterus may be capable of infecting cells that mediate important functions throughout pregnancy and infflict injuries that could compromise their functions. Thus, the underlying hypothesis here is; C. trachomatis infection of trophoblast inflicts sufficient injury that impairs trophoblast endocrine functions. We have tested this hypothesis by: assessing the status of C. trachomatis infection of trophoblast, investigating the impact of productive infection on the capacity of trophoblast to synthesize cholesterol (the precursor molecule for estrogen and progesterone biosynthesis), examining the synthesis of these steroid hormones and determined whether infection affected the production of human chorionic gonadotropin (hCG), which has a regulatory role on early trophoblast functions. We report our findings that provide mechanistic insights into the way and manner in which C. trachomatis inflicts pathologies that affect pregnancy outcome.
2. Materials and Methods
Chemicals
All chemicals and reagents were purchased from Sigma Chemical Company (St. Louis, MO) unless otherwise stated.
Trophoblast Culture
Human trophoblast cell line (JAR) (ATCC, Maryland, USA) was grown in RPMI 1640 medium with 2mM L-glutamine that is modified to contain 10mM HEPES, 1mM sodium pyruvate, 4.5g/l glucose, 1.5g/l bicarbonate (Invitrogen, Life Technologies, Carlsbad, CA) supplemented with 10% FBS (Hyclone, Logan, Utah, USA), 50μg/ml vancomycin, 10μg/ml gentamicin maintained in a 37°C, 5% CO2 humidified incubator. At subconfluency, residual medium was removed and cells were rinsed free of medium using PBS, then 2-3ml of 0.25% (w/v) Trypsin-0.53 mM EDTA solution was added to flask and observed for cell layer dispersal (usually 5min). Eight milliliter of medium was added to flask and cells were mixed by gentle pipetting. Appropriate aliquots of cell suspension were seeded in new culture vessels or into wells for experiments. Cells were tested for mycoplasma contamination periodically by staining with 4,6 diamine-2-phenyl indole dihydrochloride (Boehringer, Mannheim, Germany).
Chlamydia trachomatis culture
Chlamydia trachomatis (D serovar) was obtained from ATCC and propagated in HEp-2 cell monolayer by centrifugation (1864 X g Sorvall RC5C, SH-3000 rotor) driven infection for 1 hour followed by rocking in a humidified incubator at 37°C and 5% CO2 for 1hr 30min. The residual medium was aspirated and replaced with fresh growth medium containing FBS prescreened for chlamydia antibodies and 2μg/ml cycloheximide (cycloheximide was not used in instances where chlamydia infection was for experimental purposes). It was then returned to the humidified incubator at 37°C and 5% CO2 for 72hr. At the end of 72hr, C. trachomatis was harvested , sonicated, loaded onto discontinuous gradient of urografin (Schering, Berlin, Germany), and elementary bodies(EBs) were pelleted at 17,211 x g (Sorvall RC5C, SS-34 Rotor) for 1hr at 4°C. Harvested EBs were stored at -80°C in sucrose phosphate glutamate buffer (0.22M sucrose, 10mM sodium diphosphate, 5mM glutamic acid, pH 7.4) in small aliquots and thawed as needed [18]. C. trachomatis inclusion forming units (IFU) were determined by thawing a frozen aliquot of the harvested purified EBs and infecting confluent (5x105 cells/well) HEp-2 cell monolayers in 24 well plate with 10 fold serial dilution in medium using the centrifugation assisted procedure already described. Infected cells treated with cycloheximide were incubated at 37°C for 72 hours, washed, fixed in methanol, and stained using fluorophore labeled anti-lipopolysaccharide antibody (chlamydia identification kit, Bio-Rad, Woodinville, WA, USA). The total inclusion forming units was enumerated by counting 10 microscope fields(x 200 magnification) using an inverted fluorescent microscope (Olympus, Melville, NY, USA).
Infection of trophoblast with Chlamydia trachomatis
Trophoblast monolayer was washed with phosphate buffered saline (PBS), then infected with 1ml (6 well plate) of multiplicity of infection (MOI) of 3 elementary bodies (EBs) per cell. The capacity of C. trachomatis to infect trophoblast was assessed using similar methods described above for quantification of IFU/ml in 24 well plate but in this instance photomicrography was recorded and compared with records for the infection of HEp-2 cells using EBs from stocks in our lab at MOI=3/cell.
Assessment of Chlamydia trachomatis status in trophoblast
The efficacy of C. trachomatis infection of trophoblast was assessed by (i) determining the capacity of C. trachomatis to initiate productive and transferable infection in trophoblast. This involved the time course harvesting and purifying of EBs from infected trophoblast and titration onto HEp-2 cell monolayer (19). Chlamydial inclusion forming units (IFUs) was enumerated and compared with IFUs obtained from direct HEp-2 cells infection using EBs from our lab stock. In all instances infection was established in 25-45% of HEp-2 cells using C. trachomatis harvested from infected trophoblast. (ii) Also determining the percent infectivity of trophoblast by C. trachomatis, by counting cells in ten fields and enumerating the numbers of infected trophoblast. The time course percent infectivity was recorded and compared with time course percent infectivity of HEp-2 cells by EBs from lab stock. In some instances also, the capacity of C. trachomatis to assume a chronic course in trophoblast was assessed using accepted molecular indices by assay of chlamydial HSP-60 and MOMP protein from C. trachomatis harvested from trophoblast.
Trophoblast cholesterol assay
Trophoblast cellular cholesterol was estimated using fluorimetric procedures described in assay kit manual (Amplex Red Cholesterol Assay Kit, Molecular probes, Eugene, OR) [20]. Protein estimation [21] was also done on lysate. Cholesterol was reported as µg/mg protein.
Trophoblast estradiol assay
Trophoblast cellular estradiol was assayed using enzyme immunoassay procedures described in assay kit manual (Cayman Chemical Company, Ann Arbor, MI) [22]. Absorbance was recorded at wavelength of 415nm using microplate reader (BioRad Microplate Reader 3550, Hercules, CA). Protein estimation [21] was also done on lysate. Estradiol was reported as ng/mg protein.
Trophoblast progesterone assay
Trophoblast cellular progesterone was assayed using the fluorimetric progesterone receptor competitive assay procedure described in the assay kit manual (Progesterone Receptor Competitive Assay, Green, Invitrogen Corporation, Carlsbad, CA). In the test, glutathione transferase anchors human progesterone receptor to expose the ligand binding sites. A competitive assay is then performed between fluormone tagged to progesterone epitope and the free progesterone in the sample or standard. Progesterone displaces fluormone tagged progesterone causing increased fluorescence. Briefly, 50µl of sample or standard was put in microtiter plate and 50µl of reaction mix (40µl PR LBD progesterone receptor, 10µl Fluormone PL Green tagged progesterone ligand, 4µl DTT, 946µl PR Screening Buffer Green). Microtiter plate was incubated at room temperature while rocking for 2h. Fluorescence was recorded at emission wavelength of 485nm and an excitation wavelength of 530nm using CytoFluor 4000 ((Applied Biosciences, Woodinville, CA). Protein estimation [21] was also done on lysate. Progesterone was reported as mg/mg protein.
Assay of HMG-CoA reductase, aromatase, adrenodoxin reductase, cHSP-60, MOMP, and hCG protein
Western blot was run on lysates of uninfected trophoblast, or C. trachomatis infected trophoblast, or C. trachomatis harvested from trophoblast after time course infection. Protein was precipitated with 10% trichloroacetic acid and resuspended in assay buffer (BioRad Laboratories, CA, USA) [23]. Briefly, 25μg protein was spotted per lane on 10% SDS-polyacrylamide gel. After electrophoresis, protein bands were electrophoretically transferred to 0.2 μM Immun-Blot PVDF membrane (BioRad Laboratories, CA, USA). To avoid non-specific binding, blocking was done with 3% blocker provided with the kit, washed and incubated with primary antibodies; anti-cHSP-60, or anti-aromatase, or anti-hCG, or anti-adrenodoxin-reductase (ABCAM Inc, Cambridge, MA), or anti-MOMP (Virostat Inc, Portland ME), or anti-HMG-CoA reductase (Upstate Biotechnology, Lake Placid, NY), or anti-actin (Santa Cruz Biotech, CA) for internal control at dilutions of 1:500 for 1h, then washed and incubated with GAx-HRP (horse radish peroxidase) (BioRad Laboratories, CA, USA) at a dilution of 1:5000 for 1h, then washed with PBST buffer [24]. Colorimetric detection was done according to manufacturers instructions. Band intensity was determined using Gel Logic 200 (Kodak, Chicago, IL)
Protein Estimation
Protein assay on each sample was done by the bicinchoninic acid-copper (II) sulphate reagent assay system [21].
Statistical Analysis
All data are expressed means ± standard error of triplicate samples. Data were analyzed by Student's t-test using difference between means of two treatments and p values of 0.05 were reported as significant. In some instances, the repeated measures analysis of variance (ANOVA) model was used for the dependent variables.
3. Results
Chlamydia trachomatis induces a productive infection in human trophoblast
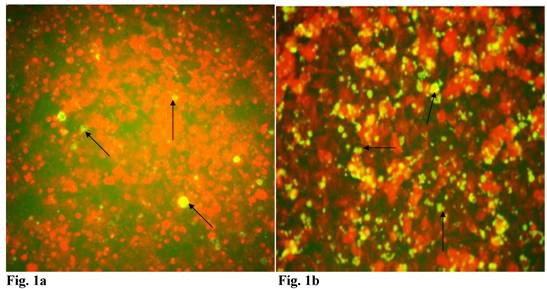
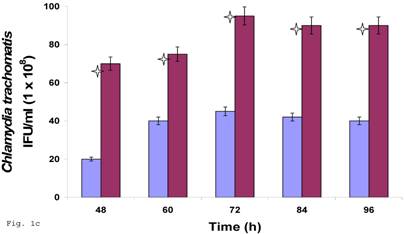
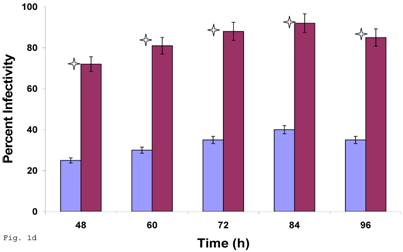
Since C. trachomatis-host interaction and the capacity of the resultant infection to impact host cell functions are dependent on transferability of infectious elementary bodies from cell to cell, we decided to investigate if it would initiate a productive infection in human trophoblast cell line, thereby providing insights into the enormity of chlamydial STD on reproductive outcome. Regular C. trachomatis inclusions were demonstrable in trophoblast (Fig. 1a), although there was significantly reduced inclusion formation when compared with results obtained in direct infection of HEp-2 cells (Fig. 1b). It is important to note that time course percent infectivity of trophoblast (direct trophoblast infection) varied from 25%-45% (Fig. 1d), a significant finding considering the nature of trophoblast. To assess the capacity of C. trachomatis to initiate productive infection in trophoblast, time course harvest of C. trachomatis EBs from trophoblast were used to infect HEp-2 cell monolayer and results showed that they exhibited efficient inclusion forming capability (Fig. 1c, grey bars). However, this result was significantly reduced (p<0.01) when compared with direct infection of HEp-2 cells (conventional cells that permit chlamydia growth) using EBs from our laboratory stock at MOI=3 EBs/cell (Fig. 1c, black bars). It is important to note that the data here are comparable to those obtained in similar experiments using macrophage cell line [19].
Induction of productive infection by Chlamydia trachomatis in trophoblast. The formation of Chlamydia trachomatis inclusion (MOI = 3/cell) in trophoblast (A) compared with direct infection of HEp-2 cells (B) is represented. Experimental procedure was repeated three times. Panel (A) indicates HEp-2 cell infection with Chlamydia trachomatis that has been harvested from trophoblast cell line JAR and titrated onto HEp-2 cells. Panel (B) shows a direct infection of HEp-2 cells with Chlamydia trachomatis. Notice the relative suppression of Chlamydia trachomatis growth in trophoblast. The arrows indicate Chlamydia trachomatis inclusion bodies. However, the Chlamydia trachomatis harvested from trophoblast were able to efficiently infect HEp-2 cells (productive infection), (C) although there is a significant difference (p < 0.01 *) in IFU/ml when Chlamydia trachomatis harvested from trophoblast are compared with regular EBs in our laboratory are used to infect HEp-2 cells (C). Further, percent infectivity of HEp-2 cells is shown in (D). There is a significant difference (p < 0.01 *) between direct infection of HEp-2 cells (red) and infection of HEp-2 cells with Chlamydia trachomatis harvested from trophoblast. All values represent means ± SEM (n = 3).
Chlamydia trachomatis exhibits increased HSP-60 shedding during infection of human trophoblast
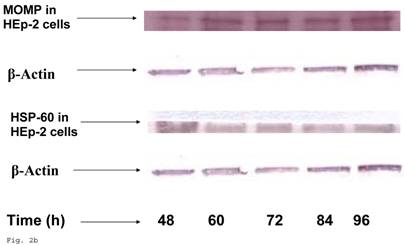
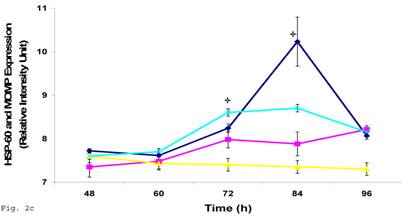
In order to evaluate the impact of infection of trophoblast on C. trachomatis forms and status, we assessed the expression of the molecular determinants of chronicity such as heat shock protein-60 (HSP-60) protein in relation to major outer membrane protein (MOMP) protein. Chlamydia trachomatis exhibited time course increase in expression of HSP-60 protein in infected trophoblast compared with infected HEp-2 cells (p<0.05), there was a more significant cHSP-60 shedding in infected trophoblast at time points after 72hr (p<0.01) with a decline at 96 h likely due to the effect of the clustering of trophoblast cells which might have impacted C. trachomatis propagation (Figs. 2a, 2b). It is important to note that experiments were set up without cycloheximide (which could annul the clustering of trophoblast at appropriate concentration – 0.3µg/ml) to avoid possible effect of drug on results. The increased expression of HSP-60 is significant, since in normal circumstances an increase in MOMP is expected with increase in infection forming units (IFUs) while HSP-60 level is suppose to be constant (see Fig. 2c in the case of HEp-2 cells).
HSP-60 shedding by Chlamydia trachomatis during infection of trophoblast. The shedding of Chlamydia trachomatis HSP-60 compared with MOMP during infection of trophoblast is depicted (A). There is time-course increase in Chlamydia trachomatis HSP-60 protein (-■-) with a peak at 72 h (B) compared with MOMP (-♦-) (p < 0.01 * ). Additionally, Chlamydia trachomatis HSP-60 shedding and MOMP expression is also shown in direct infection of HEp-2 cells (C). HSP-60 expression slowly decreases over time (-▲-), whereas MOMP (-x-) expression shows a time-course increase to 84 h before declining at 96 h (p < 0.05 *). All values represent means ± SEM (n = 3).

Chlamydia trachomatis induces an impairment of cholesterol biosynthesis in human trophoblast
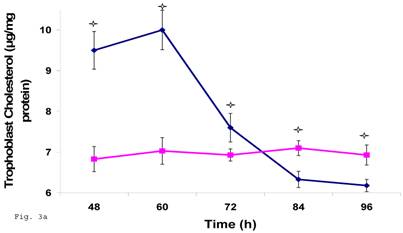
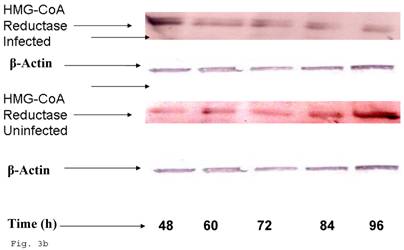
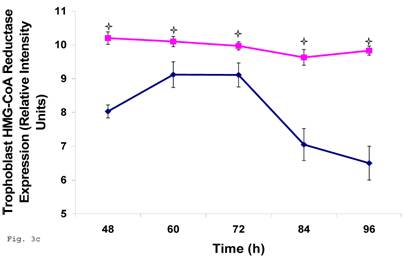
Since trophoblast play important physiologic role in the process of steroid hormone regulated implantation and placentation during pregnancy, we decided to explore the consequence of C. trachomatis infection on trophoblast capacity to synthesize cholesterol, the precursor of steroid hormones. Figure 3a shows that after an initial significant up regulation of cholesterol (p < 0.01), there was a decline below cellular cholesterol levels of uninfected trophoblast at 84 and 96 h (p<0.06). This modulation of cellular cholesterol by infection was further investigated by evaluating the effect of this event on the rate limiting enzyme of cholesterol biosynthesis, 3-hydroxy-3-methylglutaryl-Co enzyme-A (HMG-CoA) reductase. Infection down regulated the expression of HMG-CoA reductase protein expression (Figs. 3b and 3c) (p < 0.05).
Chlamydia trachomatis induces changes in cholesterol biosynthesis. The levels of cellular cholesterol in infected trophoblast (-♦-) compared with uninfected (-■-) trophoblast is depicted. There was an initial increase in cholesterol level which was significant (p < 0.05 *) and was followed by a decline (A). Protein expression (B & C) of HMG-CoA Reductase (the rate-limiting enzyme of cholesterol biosynthesis) was decreased in infected trophoblasts (-♦-) compared with uninfected trophoblasts (-■-) (p < 0.05 *). All values represent means ± SEM (n = 3).
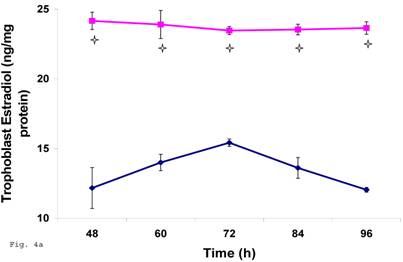
Chlamydia trachomatis infection of trophoblast down regulated estrogen biosynthesis
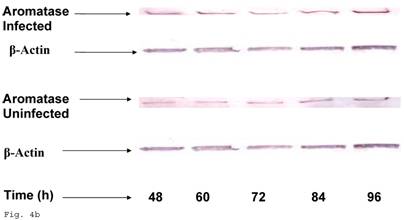
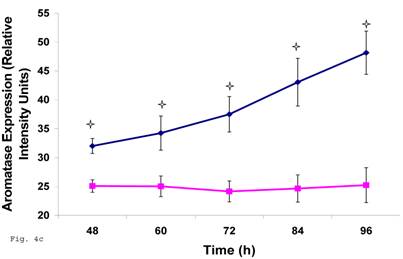
The pattern of modulation of trophoblast cholesterol biosynthesis suggests a probable accompanying interference with steroid hormone synthesis. To assess if cholesterol synthesis impairment had effect on estrogen production by trophoblast infected with C. trachomatis, we estimated the cellular estradiol and evaluated the protein of the rate limiting enzyme, aromatase. Figure 4a shows a significant decline in infected trophoblast estradiol (p < 0.01) compared with uninfected trophoblast. However, there was an up regulation of aromatase protein (Figs. 4b and 4c) (p < 0.05).
Induction of estradiol down-regulation in Chlamydia trachomatis infected trophoblast. The cellular estradiol level of infected trophoblast (-♦-) compared with uninfected cells (-■-) is depicted (A). There was significant decline in estradiol production (p < 0.01 * ) in infected trophoblast. The time-course level of aromatase production is represented in B & C. There was a significant increase in expression of the enzyme (p < 0.05 *) in infected trophoblast (-♦-) compared with uninfected trophoblast (-■-). All values represent means ± SEM (n = 3).
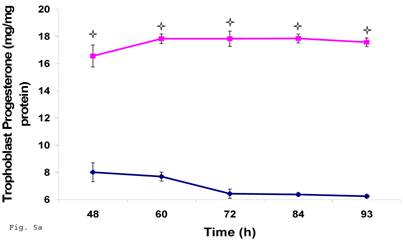
Trophoblast infected with Chlamydia trachomatis showed an impairment of progesterone biosynthesis
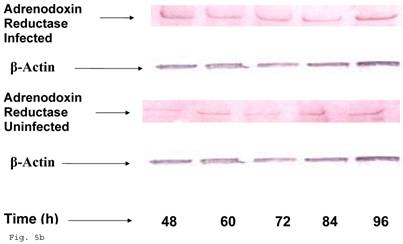
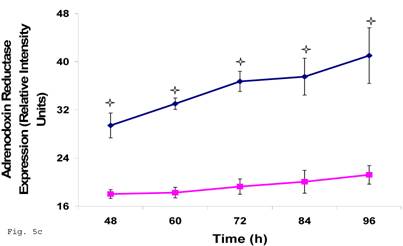
Preceding data indicate a physiologic compromise in the synthesis of trophoblast cellular cholesterol and an accompanying impact on estradiol; therefore we reasoned that additional insights could be obtained by investigating the impact of impaired trophoblast cholesterol synthesis on progesterone production. There was a decrease in cellular progesterone in C. trachomatis infected trophoblast (Fig. 5a) (p < 0.01). To further investigate what this entails in terms of the biosynthesis of progesterone, we decided to measure the protein expression of the rate limiting enzyme of progesterone biosynthesis, adrenodoxin reductase. This finding suggest that there was a possible compensatory feedback up regulation of adrenodoxin reductase protein (Figs. 5b and 5c), but the final effect of depleted cholesterol biosynthesis after 72 h may generate an impairment of substrate availability for progesterone biosynthesis.
Induction of Progesterone down-regulation in Chlamydia trachomatis infected trophoblast. The level of progesterone in trophoblast infected with Chlamydia trachomatis is shown in A. There was a significant (p < 0.01 *) decline in progesterone production in Chlamydia trachomatis infected cells (-♦-) compared with uninfected cells (-■-). The levels of adrenodoxin reductase are depicted in B & C. There was a significant (p < 0.01 *) up-regulation of enzyme in infected cells (-♦-) compared with uninfected cells (-■-). All values represent means ± SEM (n = 3).
Defective production of human chorionic gonadotropin by Chlamydia trachomatis infected trophoblast
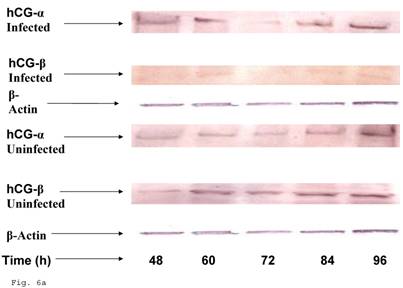
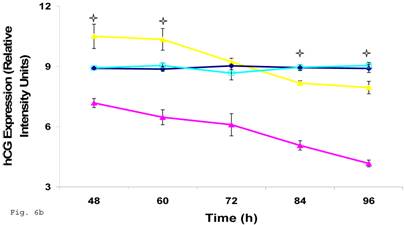
Since the induction of trophoblast function during pregnancy depends on human chorionic gonadotropin by trophoblast, we decided to assess the effect of C. trachomatis infection on hCG production by trophoblast. β-Human chorionic gonadotropin protein component of hCG was significantly depleted in C. trachomatis infected trophoblast (Figs. 6a and 6b) compared with uninfected (p < 0.05). The α-hCG protein component showed an initial increase accompanied by a decline.
Down-regulation of hCG in Chlamydia trachomatis infected trophoblast. The effect of Chlamydia trachomatis on trophoblast hCG production during infection is depicted as hCG protein expression (A and B). Panel A shows reduction in β-hCG in infected trophoblast compared with uninfected cells. Less change was recorded in α-hCG. There was a significant decline (p < 0.05 *) in β-hCG production (B) in infected trophoblast (-x-) compared with uninfected (-■-) while the α-hCG showed no significant difference when Chlamydia trachomatis infected trophoblast (-▲-) is compared with uninfected trophoblast (-♦-). All values represent means ± SEM (n = 3).
4. Discussions
Mechanistic insights into the ways and manners in which C. trachomatis inflict serious diseases, especially those affecting pregnancy outcome are needed for improved management of female reproductive life. Chlamydia trachomatis, like other chlamydiae, is known to uniquely take a chronic course, an event which is conceivably required for the protracted host-pathogen interaction and thus the establishment of pathology. Such pathology initiation has been associated with inflammatory damages that have been consistently correlated with seropositivity of chlamydial HSP-60 (cHSP-60) [25], a finding which extends to other chlamydia species [24].
There is compelling molecular evidence provided by the data reported here suggesting that C. trachomatis may assumes a chronic course in trophoblast (Figs. 2a, 2b and 2c) alongside with a relative productive infection (Fig. 1c, grey bar). Arguably this event is less significant compared with direct infection of HEp-2 cell (Fig. 1c, black bar), which are more conventional cells for chlamydia propagation. This finding is important. First, trophoblast cells are avidly phagocytic and are endowed with the capacity to produce reactive nitrogen and oxygen intermediates and other lethal biomolecules [26, 27, 28], re-enforcing the hypothesis of a protective role for trophoblast against infectious agents at the fetal-maternal interface [3]. This characteristic of trophoblast should render it non-susceptible to C. trachomatis. Despite this feature of trophoblast, C. trachomatis is able to colonize it and produce transferable infection. Therefore it stands to reason that the endowment of trophoblast with capacity to evoke such immune defenses may account for the differences in infectivity of trophoblast compared with HEp-2 cells. Second, the up regulation of cHSP-60 shedding, which is suggestive of chronicity [29], implies that such infection may not be transient and arguably impacts the functional capabilities of trophoblast, especially steroid hormone biosynthesis; activities that are of tremendous importance for fetal-maternal relation. It is important to note that the observations reported here are changes observed in infected trophoblast cell line JAR and not primary trophoblast cells which may produce different pattern of responses following chlamydia infection. The possibility of replicating these findings is the basis of an on-going in vivo study in our laboratory.
Chlamydia trachomatis uniquely harbors eukaryotic host cell cholesterol in its EBs and parasitophorous vacuole membrane, with evidence that such biomolecules are trafficked from host system [30]. Previous reports [23] have provided compelling evidence that in chlamydial infectious course in macrophage (forms suggestive of chronicity), the impact of cholesterol trafficking from host cell to chlamydia results in depleted host cell cholesterol; thus starving host cell of cholesterol for other requirements such as membrane biosynthesis. We report here a finding of an initial increase in trophoblast cellular cholesterol with an accompanying decline (Fig. 3a), which we reasoned may have impacted host cellular biosynthesis of steroid hormones (estrogen and progesterone). The levels of estrogen and progesterone are critical factors in pregnancy sustenance [31]. They enrich the uterus with thick lining of blood vessels and capillaries so that it can sustain the growth of fetus. We report a decline in estrogen and progesterone in chlamydial infected trophoblast and an accompanying positive feedback up regulation in the rate limiting enzymes in the biosynthetic pathways of these hormones. However, such up regulation of enzymes did not manifest in hormone production because of starvation in substrate (cholesterol) levels. This report of impaired steroid hormone metabolism is important, especially so in view of the fact that invitro studies abound demonstrating a correlation between changes in steroid hormones metabolism, steroid hormone receptor expression and the event of implantation and placentation [32].
Trophoblast impaired estrogen and progesterone production can account for failure of trophoblast invasion of the endometrium and has been associated with some cases of pre-eclampsia [33, 34, 35]. The importance of this finding about compromised estrogen and progesterone biosynthesis in C. trachomatis infected trophoblast is of enormous significance in the face of previous reports of unexplainable induction of abortion by chlamydia [17]. Human chorionic gonadotropin is not only the regulator of trophoblast steroid hormone biosynthesis; it also acts synergistically alongside other factors from the ovary to establish a receptive endometrium [36]. We found a decline in β-hCG protein in infected trophoblast (Fig. 6b). The reason for the decline is not very clear, however, it is important to note that the cysteine requirement of C. trachomatis MOMP is enormous and infected trophoblast may have to competitively channel cysteine to MOMP and hCG (which also has a lot of cysteine amino acid units in its β-hCG sequence), thus compromising the level of amino acid available.
This study has provided some mechanistic insights into the physiologic change meted on trophoblast by C. trachomatis infection which through the establishment of chronic onset along with relative productive infection depletes cholesterol and hCG elaboration. These findings of impaired trophoblast functions provide additional details in line those reported by Equils et al., 2006 [37], in which cHSP-60 was used to mediate trophoblast apoptosis. Also, it provides further understanding on how C. trachomatis plays an etiologic role in pathogenesis of disturbed pregnancies, other than the classical explanation of possible fibrosis of fallopian tubes and infection of newborn. The physiologic events reported in our study shows the dynamics of biomolecular trafficking between human trophoblast and intracellular C. trachomatis, as well as their effects on biosynthetic processes and a predictable probable impairment of proper pregnancy development.
Acknowledgements
This work was supported by funds made available in the form of the Shaw Scientist Award to A.A.A. by the Greater Milwaukee Foundation.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Bevilacqua E, Abahamsohn P.A. Ultrastructure of trophoblast giant cell transformation during the invasive stage of implantation of the mouse embryo. J. Morphology. 1988;198:341-351
2. Mehrotra P.K. Ultrastructure of mouse ectoplacental cone cells. Biol. Struct. Morph. 1988;1:63-68
3. Kanai-Azuma M, Kanai Y, Kurohmaru M, Tachi C, Yazaki K, Hayashi Y. Giant cells transformation of trophoblast cells in mice. Endocrine J. 1994;41:33-41
4. Loke YW, King A. Human Implantation. Cambridge: Cambridge University Press. 1995
5. Manyonda I.T, Slater D.M, Fenske C, Hole D, Choy M.Y, Wilson C. A role for noradrenaline in pre-eclampsia: towards a unifying hypothesis for the pathophysiology. Br. J. Obstet. Gynaecol. 1998;105:641-648
6. Katsuragawa H, Rote N.S, Inoue T, Narukawa S, Kanzaki H, Mori T. Monoclonal antiphosphatidylserine antibody reactivity against human first-trimester placental trophoblasts. Am. J. Obstet. Gynecol. 1995;172:1592-1597
7. Sugimura M, Kobayashi T, Shu F, Kanayama N, Terao T. Annexin V inhibits phosphatidylserine-induced intrauterine growth restriction in mice. Placenta. 1999;20:555-560
8. Kanenishi K, Kuwabara H, Ueno M, Sakamoto H, Hata T. Immunohistochemical adrenomedullin expression is decreased in the placenta from pregnancies with pre-eclampsia. Pathol. Int. 2000;50:536-540
9. Page N.M, Woods R.J, Gardiner S.M, Lomthaisong K, Gladwell R.T, Butlin D.J. et al. Excessive placental secretion of neurokinin B during the third trimester causes pre-eclampsia. Nature. 2000;405:797-800
10. Holland M.J, Bailey R.L, Hayes L.J, Whittle H.C, Mabey D.C.W. Conjunctival scarring in trachoma is associated with depressed cell-mediated immune responses to chlamydial antigens. J. Infect. Dis. 1993;168:1528-1531
11. Marais N.F, Wessels P.H, Smith M.S, Gericke G.A. Prevalence of Chlamydia trachomatis infection in new patients at the infertility clinic. S. Afr. Med. J. 1990;77:232-233
12. Chow J.N, Yenekura N.L, Richwald G.A, Greenland S, Sweet R.L, Schachter J. The association between Chlamydia trachomatis and ectopic pregnancy. A matched-pair, case-control study. JAMA. 1990;263:3191-3192
13. Beatty W.L, Morrison R.P, Byrne G.L. Persistent chlamydiae: from cell culture to a paradigm for chlamydial pathogenesis. Microbiol. Rev. 1994;58:686-699
14. Macauley M.E, Riordan T, James J.M, Laventhall P.A, Morris E.M, Neal B.R. et al. A prospective study of genital infections in a family planning clinic. Epidemiol. Infect. 1990;104:55-61
15. Azenabor A.A, Eghafona N.O. Association of Chlamydia trachomatis antibodies with genital contact disease in women in Benin City, Nigeria. Tropical Medicine and International Health. 1997;2:389-392
16. Roberts W, Grist N.R, Giroud P. Human abortion associated with infection by ovine abortion agent. Br. Med. J. 1967;4:37
17. Johnson F.W.A, Matheson B.A Williams H, Laing A.G Jandial V, Davidson-Lamb R Halliday G.J, Hobson D Wong S.Y, Hadley K.M Moffat, M.A.J Postlethwaite R. Abortion due to infection with Chlamydia psittaci in a sheep farmer's wife. Br. Med. J. 1985;290:592-594
18. Caldwell H.D, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect. Immun. 1981;31:1161-1176
19. Azenabor A.A, Chaudhry A.U. Chlamydia pneumoniae survival in macrophages is regulated by free Ca2+ dependent reactive nitrogen and oxygen species. J. Infect. 2003;46:120-128
20. Amundson D.M, Zhou M. Fluorimetric method for the enzymatic determination of cholesterol. J. Biochem. Biophys. Methods. 1999;38:43-52
21. Smith P.K, Krohn R.I, Hermanson G.T, Mallia A.K, Gartner F.H, Provenzano M.D, Fujimoto E.K, Goeke N.M, Olson B.J, Klenk D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76-85
22. Erickson G.F. The ovary: basic principles and concepts. A. physiology. In: (ed.) Felig P, Baxter J.D, Frohman L.A. Endocrinology and metabolism. New York: McGraw-Hill. 1995:973-1015
23. Azenabor A.A, Job G, Adedokun O.O. Chlamydia pneumoniae infected macrophages exhibit enhanced plasma membrane fluidity and show increased adherence to endothelial cells. Mol. and Cell. Biochem. 2005;269:69-84
24. Azenabor A.A, Muili K, Akoachere J.F, Chaudhry A. Macrophage antioxidant enzymes regulate Chlamydia pneumoniae chronicity: Evidence of redox balance on host-pathogen relationship. Immunobiol. 2006;211:325-329
25. Morrison R.P. Chlamydial hsp60 and the immunopathogenesis of chlamydial disease. Seminars in Immunology. 1991;3:25-33
26. Gagioti S, Colepicolo P, Bevilacqua E. Post implantation mouse embryos have the capability to generate and release reactive oxygen species. Reprod. Fertil. Dev. 1995;7:1111-1116
27. Gagioti S, Colepicolo P, Bevilacqua E. Reactive oxygen species and the phagocytosis process of hemochorial trophoblast. Ciencia e Cultura. 1996;48:37-42
28. Gagioti S, Scavone C, Bevilacqua E. Participation of the mouse implanting trophoblast in nitric oxide production during pregnancy. Biol. Reprod. 2000;62:260-268
29. Azenabor A.A, Chaudhry A.U. and Yang S. Macrophage L-type Ca2+ channel antagonists alter Chlamydia pneumoniae MOMP and HSP-60 mRNA gene expression, and improve antibiotic susceptibility. Immunobiol. 2003;207:237-245
30. Carabeo R.A, Mead D.J, Hackstadt T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc. Natl. Acad. Sci. 2003;100:6771-6776
31. Kam E.P.Y, Gardner L Loke Y.W, King A . The role of trophoblast in the physiological change in decidual spiral arteries. Hum. Reprod. 1999;14:2131-2138
32. Lunghi L, Ferretti M.E, Medici S, Biondi C, Vesce F. Control of human trophoblast function. Reprod. Biol. Endocrinol. 2007;5:6
33. Khong T.Y, de Wolf F, Robertson W.B, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br. J. Obstet. Gyanaecol. 1986;93:1049-1059
34. Pridjian G, Puschett J.B. Preeclampsia. Part 1: clinical and pathophysiologic considerations. Obstet Gynecol Surv. 2002;57:598-618
35. Pridjian G, Puschett J.B. Preeclampsia. Part 2: experimental and genetic considerations. Obstet Gynecol Surv. 2002;57:619-640
36. Reshef E. Lei Z. M, Rao C.V, Pridham D.D, Chegini N, and Luborsky J.L. The presence of gonadotropin receptors in nonpregnant human uterus, human placenta, fetal membranes and decidua. J. Clin. Endocrinol. Metab. 1990;70:421-430
37. Equils O, Lu D, Gatter M, Witkin S.S, Bertolotto C, Arditi M, McGregor J.A, Simmons C.F, Hobel C.J. Chlamydial Heat Shock Protein 60 Induces Trophoblast Apoptosis through TLR4. J. Immunol. 2006;177:1257-1263
Author contact
![]() Correspondence to: Dr. Anthony A. Azenabor, Enderis Hall, Room 469, University of Wisconsin, 2400 E. Hartford Avenue, Milwaukee, WI 53211 USA. Phone: (414) 229-5637; Fax: (414) 229-2619; Email: aazenaboedu
Correspondence to: Dr. Anthony A. Azenabor, Enderis Hall, Room 469, University of Wisconsin, 2400 E. Hartford Avenue, Milwaukee, WI 53211 USA. Phone: (414) 229-5637; Fax: (414) 229-2619; Email: aazenaboedu