Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2007; 4(3):140-145. doi:10.7150/ijms.4.140 This issue Cite
Research Paper
BRCA1 May Modulate Neuronal Cell Cycle Re-Entry in Alzheimer Disease
Teresa A. Evans1, Arun K. Raina1, André Delacourte2, Olga Aprelikova3, Hyoung-gon Lee1, Xiongwei Zhu1, George Perry1,4, Mark A. Smith1 ![]()
1. Department of Pathology, Case Western Reserve University, Cleveland, Ohio 44106, USA
2. Inserm U837, JPARC, Bat. G. Biserte, 1 place de Verdun, 59045 Lille cedex, France
3. Laboratory of Biosystems and Cancer, National Cancer Institute, NIH, Bethesda, Maryland 20892, USA
4. College of Sciences, University of Texas at San Antonio, San Antonio, Texas 78249, USA
Received 2007-4-11; Accepted 2007-5-9; Published 2007-5-12
Abstract
In Alzheimer disease, neuronal degeneration and the presence of neurofibrillary tangles correlate with the severity of cognitive decline. Neurofibrillary tangles contain the antigenic profile of many cell cycle markers, reflecting a re-entry into the cell cycle by affected neurons. However, while such a cell cycle re-entry phenotype is an early and consistent feature of Alzheimer disease, the mechanisms responsible for neuronal cell cycle are unclear. In this regard, given that a dysregulated cell cycle is a characteristic of cancer, we speculated that alterations in oncogenic proteins may play a role in neurodegeneration. To this end, in this study, we examined brain tissue from cases of Alzheimer disease for the presence of BRCA1, a known regulator of cell cycle, and found intense and specific localization of BRCA1 to neurofibrillary tangles, a hallmark lesion of the disease. Analysis of clinically normal aged brain tissue revealed systematically less BRCA1, and surprisingly in many cases with apparent phosphorylated tau-positive neurofibrillary tangles, BRCA1 was absent, yet BRCA1 was present in all cases of Alzheimer disease. These findings not only further define the cell cycle reentry phenotype in Alzheimer disease but also indicate that the neurofibrillary tangles which define Alzheimer disease may have a different genesis from the neurofibrillary tangles of normal aging.
Keywords: Alzheimer disease, BRCA1, cell cycle, oncogenesis
1. Introduction
Neurofibrillary tangles (NFT) are the cardinal intracellular lesion of Alzheimer disease (AD), and are also found in normal aging, albeit to a lesser extent. Highly phosphorylated tau protein is considered the predominant proteinaceous component of NFT [1], however, numerous other proteins have also been localized to these lesions including neurofilaments [2], ubiquitin [3, 4], amyloid-β [5], and cell cycle markers [6-11]. Notably, NFT associated with normal aging are viewed as being quantitatively different, but qualitatively identical [12]. Whether the mechanisms responsible for the genesis of NFT in AD are similar or different from the genesis of NFT in normal aging is unknown.
BRCA1 is expressed in dividing neuronal cells during development, and is present in smaller amounts in fully differentiated cells [13]. BRCA1 is known to regulate transcription, regulate cell cycle progression, and may even have a role in maintaining telomere function and as such the presence of BRCA1 is indicative of cell cycle changes and DNA damage, both of which are pathogenic changes in AD. Nucleic acid damage is well-documented in AD, specifically within the pyramidal neurons, the population susceptible to neurodegeneration and death [14-18]. Consequently, tumor suppressor proteins such as p21, p27, p53 are activated by BRCA1, are indicative of DNA damage [19], and are activated in AD [6, 20]. Such tumor suppressors play a role in suppression of the cell cycle and cell survival instead of apoptosis and their presence may be a neuroprotective factor to prolong the life of the cell after re-entry into the cell cycle, protecting neurons from completion of apoptosis [21]. These proteins have come to the forefront as molecular candidates to be used in discrimination between normal aging and pathological diseases. Neuroprotective factors have also been suggested as a possible target for drug design efforts with the goal of halting the progression of the cell cycle and delaying apoptosis. [22-24]. BRCA1 is also associated with a spectrum of functions related to the preservation of genomic stability [25]. For example, BRCA1 is involved in transcriptional activation and growth inhibition [26-28], transcription coupled repair (TCR) of oxidative damage to DNA and other DNA repair [29, 30], and association with γ-tubulin, a central component of the microtubule organizing center and centrosomes, thus implying a regulatory role in G2/M progression [31]. There are also a host of putative functions assigned to BRCA1 based on its structure and associations. Among these include association with BARD1, cyclin A and cyclin D kinases which phosphorylate BRCA1 [32].
Many of these known functions of BRCA have also been associated with AD. Oxidative DNA damage, as well as RNA damage [15, 16], has been well documented in the aging brain, contributing to the development of AD [18]. Further, even cases of mild cognitive impairment display the same abnormalities, prompting the search for increased DNA repair mechanism in cases of neurodegeneration [33]. Evidence of cell cycle dysfunction and the oxidative DNA damage profile in AD caused speculation that BRCA1 may play a role in disease pathogenesis.
There are clearly a number of striking parallels between AD and cancer, including age, and likely multiple etiologies and risk factors [34]. As for cancer, the notion of a “two-hit” hypothesis has also been proposed [35, 36]. The latter may separate AD from normal aging. Indeed, while cells, in this case neurons, have the capacity to maintain homeostatic balance and function under condition of stress, several “hits” may disrupt the cells' regeneration capacity leading to neurodegeneration and death. This stress may be oxidative insult or metabolic inefficiency. Possibilities for other “hits” include genetic mutations in apolipoprotein E, presenilins, or amyloid-β protein precursor, hormonal dysregulation, environmental or education status, inflammatory responses, or perhaps even the induction of oncogenic-like pathways [37]. To this end, we found that the BRCA1 protein is strongly associated with NFT in AD yet a feature of only about half of the cases of normal aging containing tau-positive NFT. Elevations in BRCA1 in neurons in AD may represent an attempt towards homeostasis by the cell, working with other factors to halt the cell cycle and mediate DNA repair. Interestingly, a much higher proportion of NFT were labeled in AD cases than in control cases. These findings hint at differential mechanisms of NFT genesis in AD and in normal aging and/or distinct cellular responses to these changes.
2. Materials and Methods
Hippocampal and cortical specimens were obtained postmortem from patients with histopathologically confirmed AD (n = 33, age 65-93, mean 82.3) and control (n = 28, age = 47-89, mean 73.5). Tissue was fixed either in 10% buffered formalin or in methacarn (methanol:chloroform:acetic acid, 6:3:1), and embedded in paraffin. 6μm sections were deparaffinized in xylene and rehydrated in graded alcohol, the endogenous peroxidase activity eliminated by incubation in 3% hydrogen peroxide in methanol for 30 min, and finally to Tris buffered saline (TBS, 50 mM Tris, 150 mM NaCl, pH=7.6). Sections were blocked in 10% normal goat serum (NGS) for 30 min followed by overnight incubation with primary antibody in 1% NGS at 4°C in a humidified chamber. Staining was completed using the peroxidase-anti-peroxidase procedure with 3,3-diaminobenzidine (DAB) as chromagen, and sections were dehydrated and mounted with permount.
Antibodies used included monoclonals recognizing BRCA1 amino acids 1-304 (Clone MS110, Oncogene Research Products), rabbit polyclonal against phosphorylated BRCA1 amino acids 1489-1500 (Upstate Cell Signaling Solutions), and phosphorylated tau (AT8, Endogen) to label NFT.
Antibody specificity for BRCA11-304 was confirmed by performing an adsorption experiment with its corresponding antigen. Diluted antibody was incubated overnight with 20μg of BRCA1 peptide and applied to an adjacent section with antibody alone. Additionally, cross-adsorption with purified tau protein was performed as well as omission of primary antibody.
To further analyze the presence of BRCA1 in cases of control, mild cognitive impairment, as well as AD, formalin fixed blinded sections were analyzed for BRCA1 and phosphorylated tau. Using images obtained with a Zeiss Axiocam and associated image analysis software, the number of NFT immunostained in 3 fields (1mm2) encompassing the CA1 and CA2 areas of the hippocampus were determined.
3. Results
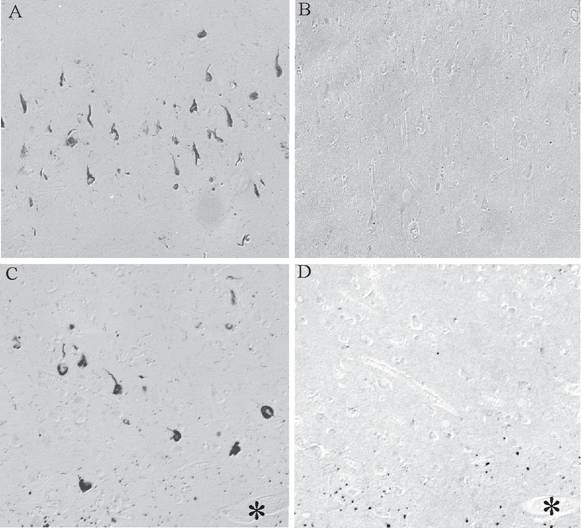
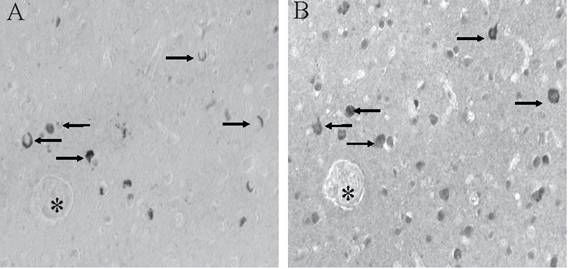
BRCA1 is found to be specifically and intensely localized with intracellular NFT in hippocampal neurons in AD (Figure 1A). In young control cases and those without any tau pathology, no cellular staining was seen (Figure 1B). The specificity of our findings was demonstrated in adjacent sections where BRCA1 immunoreactivity in NFT (Figure 1C) was completely abolished following adsorption with the specific BRCA1 peptide (Figure 1D). On the other hand, cross-adsorption with tau protein did not diminish the immunoreaction (data not shown). BRCA1 localization to NFT was detected in all cases of AD, independent of fixation methods.
Monoclonal antibody to BRCA1 recognizes intracellular NFT in all cases of AD (A), yet in many control cases, no structures are stained (B). On adjacent serial sections, the specific localization of BRCA1 to NFT (C) is completely abolished following adsorption with antigen (D). * marks landmark vessel. Scale bar= 50 μm.
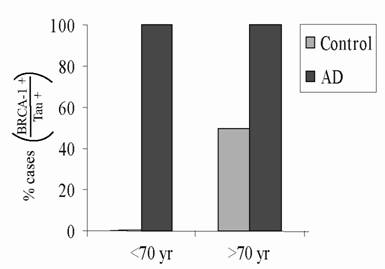
Hippocampal sections from 17 clinically normal cases containing pathological accumulations consistent with normal aging were specifically chosen and immunostained for BRCA1 and AT8. It was noted that in many of the control cases containing phosphorylated tau-positive NFT, BRCA1 was absent. Analysis of this series of cases shows that while all cases with AD exhibited BRCA1-positive NFT, BRCA1 was present to a lesser extent and in smaller and more variable numbers in control cases with pathology across all age ranges (Figure 2).
All AD cases and all clinically normal cases showing AT8-positive NFT consistent with normal aging were also analyzed for the presence of BRCA1-positive NFT. All AD cases (100%) at all age ranges exhibited BRCA1 positive NFT. Yet only about half of the control cases with NFT displayed BRCA1 positivity.
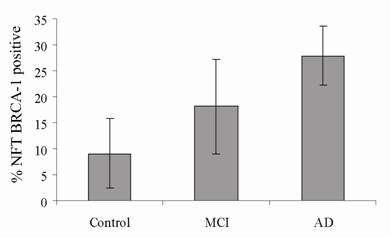
To further assess the relationship between BRCA1 and AD, blinded sections were stained for BRCA1 and AT8 in well characterized cases classified as control (no neurological diagnosis), mild cognitive impairment (MCI) and AD. The numbers of NFT stained for each marker in three fields were quantified using a computer assisted image analysis (Figure 3) and expressed as the percentage of BRCA1 positive compared to AT8 positive NFT. In control cases (n = 4, age range 83-93), an average of only 9% of NFT contained BRCA1. In cases with MCI (n = 3, age range 78-96), 18% of NFT were BRCA1 were positive, and in cases of AD (n = 3, age range 69-91), the number increased to 28%. The percentage of NFT stained in MCI cases is essentially midway between AD and control cases consistent with clinical findings that MCI is a transition. Nonetheless, as expected, by looking only at tau, there was a wide variety of pathology in each category. In the control cases, the number of tau-positive NFT ranged from 5 to 260, and in AD cases, from 39 to 321 NFT.
The number of NFT stained for BRCA1 and AT8 were counted in the CA1/CA2 regions of hippocampus in well characterized cases of AD (n = 3), MCI (n = 3), and control (n = 4). In AD, an average 28% of AT8-positive NFT contained BRCA1. That number was only 18% for cases of MCI and 9% for the control cases. While all four control cases contained AT8-positive NFT, only two cases displayed BRCA1, a finding similar to that observed with the aged controls examined in Figure 2.
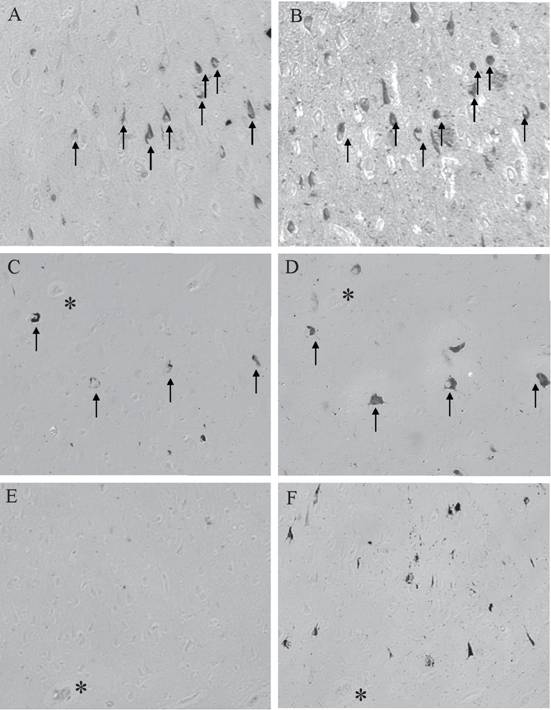
The colocalization of AT8 and BRCA1 representative of the different disease states is shown in Figure 4. Adjacent serial sections of AD showed that large numbers of NFT are positive for BRCA1 (Figure 4A) with significant overlap with AT8 (Figure 4B). In a case of MCI, while fewer AT8-positive NFT are present (Figure 4D), again there is significant overlap with BRCA1 (Figure 4C). As was seen in about half of the clinically normal cases with tau pathology, while even moderate numbers of AT8-positive NFT are present (Figure 4F), BRCA1 (Figure 4E) is not present.
The localization patterns of AT8 and BRCA1 in the different disease states varies greatly. AD cases show high numbers of AT8-positive NFT (B), with many overlapping with BRCA1 localization (A). In cases of MCI, while there are fewer AT8-positive NFT (D), many overlap with BRCA1 (C). Yet in about half of the aged control cases, while there are moderate numbers of AT8-positive NFT (F), BRCA1 is absent (E). * denotes landmark vessels on adjacent serial sections. Arrow mark NFT labeled for both BRCA1 and AT8.
Qualitative analysis for the presence of phosphorylated BRCA1 (pBRCA1) was also performed. In some cases of AD, pBRCA1 stained neuronal nuclei as well as a smaller population of NFT (Figure 5B) compared to non-phosphorylated BRCA1 (Figure 5A).
Phosphorylated BRCA1 is localized in some cases of AD to both nuclei as well as some NFT (B). In adjacent serial sections stained for BRCA1 (A), many of the cells containing NFT (arrows) also contain pBRCA1 (B). * denotes landmark vessel.
4. Discussion
In this study, we show that, controlled for age, there is a progression in the percentage of NFT containing BRCA1 from cases with no dementia to MCI to AD. MCI cases, by their definition, are in the early stages of AD. The differences in BRCA1 and tau co-localization in control versus MCI or AD cases may point to different etiologies and/or different cellular responses. Indeed, mechanisms involved in the formation of NFT in AD may be very different to the process during normal aging, i.e., that the development of AD requires two or more “hits”. Neurons can maintain normal function and combat assault from oxidative damage throughout aging, unless there is another “hit”, whether it be a genetic mutation or metabolic dysfunction, from which the cell cannot overcome and maintain balance, resulting in neuronal death. As AD is a disease that can last ten years or more, rather than succumbing to apoptosis immediately, neurons may attempt to survive by initiating cell cycle progression, and attempting to control the deregulated cell cycle and concurrent apoptotic signaling. BRCA1 is a tumor suppressor protein, involved in DNA repair, suspension of the cell cycle and probable temporary delay of apoptosis when problems are suspected. Since the prevalence of BRCA1 increases as the disease progresses, transcription of BRCA1 may be activated early in the progression of AD. This is consistent with the hypothesis that cell cycle changes take place very early in the progression of the disease, long before the presence of other pathology. Over time, DNA and cell cycle changes may compound, and BRCA1 and other protein expression increases, eventually resulting in cell death. These findings raise the possibility that BRCA1 accumulates in neurons early in the disease and only in those cases in the early stages of AD and may or may not be independent of tau formation and the expression of other cell cycle markers.
The association of BRCA1 with neurodegenerative pathology in AD implicates genomic instability and possibly a neuroprotective element in neurons in AD. The emerging evidence of genomic instability as a proximal feature in the pathogenesis of neurodegeneration in AD may possibly be a feature of cell cycle instability in neurons [38]. Taken together with the association of BRCA1, this phenotype bears many resemblances to a mitotic lesion or, at minimum, the presence of oncogenic signaling in AD, providing another driving force, or “hit” specific for lesion development in AD [36]. The presence of BRCA1 and other tumor suppressor proteins is also indicative of protective mechanisms against the formation of a cancer or unnecessary apoptosis.
The presence of phosphorylated BRCA1 has been characterized under conditions of DNA damage. Phosphorylation and changes in subcellular localization follow DNA damage in cell models. For instance, phosphorylation of specific residues dictate both localization and function [39], which could be related to the varying nuclear accumulations seen in the brain in the present study. pBRCA1 has also been implicated to play a role in maintaining genomic integrity in mitochondria and in the nucleus [40]. Recent work has related these functions specifically to telomere maintenance. In BRCA1 -/- cells, telomere dysfunction evidenced by a loss of telomere repeats was found [41], a distinctive feature of degenerating neurons in the AD brain [42].
While the mechanisms responsible for the localization of BRCA1 to NFT remain to be determined, one intriguing hypothesis is that the presence of BRCA1 signifies a neurogenic/oncogenic stimulus that is found in AD and other neuropathology. In this regard, there are several examples showing cognitive improvements in dementia patients undergoing chemotherapy [43]. It would be interesting to investigate the therapeutic efficacy of combination or simply agent antimitotic therapy with vincristine, carmustine, melphalan, cyclophosphamide, or prednisone for AD [44].
Acknowledgements
Work in the authors' laboratory is support by the National Institutes of Health, the Alzheimer's Association, and by Philip Morris USA Inc. and Philip Morris International. OA was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Grundke-Iqbal I, Iqbal K, Tung YC. et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913-7
2. Perry G, Rizzuto N, Autilio-Gambetti L. et al. Paired helical filaments from Alzheimer disease patients contain cytoskeletal components. Proc Natl Acad Sci U S A. 1985;82:3916-20
3. Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641-4
4. Perry G, Friedman R, Shaw G. et al. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci U S A. 1987;84:3033-6
5. Perry G, Cras P, Siedlak SL. et al. Beta protein immunoreactivity is found in the majority of neurofibrillary tangles of Alzheimer's disease. Am J Pathol. 1992;140:283-90
6. Ogawa O, Lee HG, Zhu X. et al. Increased p27, an essential component of cell cycle control, in Alzheimer's disease. Aging Cell. 2003;2:105-10
7. Ogawa O, Zhu X, Lee HG. et al. Ectopic localization of phosphorylated histone H3 in Alzheimer's disease: a mitotic catastrophe? Acta Neuropathol (Berl). 2003;105:524-8
8. McShea A, Harris PL, Webster KR. et al. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933-9
9. Nagy Z, Esiri MM, Cato AM. et al. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl). 1997;94:6-15
10. Nagy Z, Esiri MM, Smith AD. Expression of cell division markers in the hippocampus in Alzheimer's disease and other neurodegenerative conditions. Acta Neuropathol (Berl). 1997;93:294-300
11. Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol. 1996;132:413-25
12. Castellani RJ, Lee HG, Zhu X. et al. Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol (Berl). 2006;111:503-9
13. Korhonen L, Brannvall K, Skoglosa Y. et al. Tumor suppressor gene BRCA-1 is expressed by embryonic and adult neural stem cells and involved in cell proliferation. J Neurosci Res. 2003;71:769-76
14. Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71:2034-40
15. Nunomura A, Perry G, Pappolla MA. et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959-64
16. Nunomura A, Perry G, Aliev G. et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759-67
17. Su JH, Deng G, Cotman CW. Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer's disease brain. Brain Res. 1997;774:193-9
18. Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994;36:747-51
19. Somasundaram K. Breast cancer gene 1 (BRCA1): role in cell cycle regulation and DNA repair--perhaps through transcription. J Cell Biochem. 2003;88:1084-91
20. Gartner U, Holzer M, Arendt T. Elevated expression of p21ras is an early event in Alzheimer's disease and precedes neurofibrillary degeneration. Neuroscience. 1999;91:1-5
21. Raina AK, Hochman A, Zhu X. et al. Abortive apoptosis in Alzheimer's disease. Acta Neuropathol (Berl). 2001;101:305-10
22. Suzuki A, Tsutomi Y, Akahane K. et al. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17:931-9
23. Eymin B, Sordet O, Droin N. et al. Caspase-induced proteolysis of the cyclin-dependent kinase inhibitor p27Kip1 mediates its anti-apoptotic activity. Oncogene. 1999;18:4839-47
24. Blagosklonny MV. Are p27 and p21 cytoplasmic oncoproteins? Cell Cycle. 2002;1:391-3
25. Rahman N, Stratton MR. The genetics of breast cancer susceptibility. Annu Rev Genet. 1998;32:95-121
26. Chapman MS, Verma IM. Transcriptional activation by BRCA1. Nature. 1996;382:678-9
27. Holt JT, Thompson ME, Szabo C. et al. Growth retardation and tumour inhibition by BRCA1. Nat Genet. 1996;12:298-302
28. Monteiro AN, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci U S A. 1996;93:13595-9
29. Gowen LC, Avrutskaya AV, Latour AM. et al. BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science. 1998;281:1009-12
30. Scully R, Chen J, Ochs RL. et al. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425-35
31. Hsu LC, White RL. BRCA1 is associated with the centrosome during mitosis. Proc Natl Acad Sci U S A. 1998;95:12983-8
32. Chen Y, Chen CF, Riley DJ. et al. Aberrant subcellular localization of BRCA1 in breast cancer. Science. 1995;270:789-91
33. Mecocci P. Oxidative stress in mild cognitive impairment and Alzheimer disease: a continuum. J Alzheimers Dis. 2004;6:159-63
34. Raina AK, Garrett MR, Previll LA. et al. Oncogenic parallels in Alzheimer disease. Int J Neuroprotec Neuroregen. 2006;2:80-5
35. Zhu X, Castellani RJ, Takeda A. et al. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the 'two hit' hypothesis. Mech Ageing Dev. 2001;123:39-46
36. Zhu X, Raina AK, Perry G. et al. Alzheimer's disease: the two-hit hypothesis. Lancet Neurol. 2004;3:219-26
37. Zhu X, Lee HG, Perry G. et al. Alzheimer disease, the two-hit hypothesis: An update. Biochim Biophys Acta. 2007;1772:494-502
38. Webber KM, Raina AK, Marlatt MW. et al. The cell cycle in Alzheimer disease: a unique target for neuropharmacology. Mech Ageing Dev. 2005;126:1019-25
39. Okada S, Ouchi T. Cell cycle differences in DNA damage-induced BRCA1 phosphorylation affect its subcellular localization. J Biol Chem. 2003;278:2015-20
40. Coene ED, Hollinshead MS, Waeytens AA. et al. Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol Biol Cell. 2005;16:997-1010
41. McPherson JP, Hande MP, Poonepalli A. et al. A role for Brca1 in chromosome end maintenance. Hum Mol Genet. 2006;15:831-8
42. Franco S, Blasco MA, Siedlak SL. et al. Telomeres and telomerase in Alzheimer's disease: epiphenomena or a new focus for therapeutic strategy? Alzheimers Dementia. 2006;2:164-8
43. Keimowitz RM. Dementia improvement with cytotoxic chemotherapy. A case of Alzheimer disease and multiple myeloma. Arch Neurol. 1997;54:485-8
44. Woods J, Snape M, Smith MA. The cell cycle hypothesis of Alzheimer's disease: Suggestions for drug development. Biochim Biophys Acta. 2007;1772:503-8
Author contact
![]() Correspondence to: Mark A. Smith, Ph.D., Department of Pathology, Case Western Reserve University, 2103 Cornell Road, Cleveland, Ohio 44106 USA. Tel: 216-368-3670, Fax: 216-368-8964, mark.smithedu
Correspondence to: Mark A. Smith, Ph.D., Department of Pathology, Case Western Reserve University, 2103 Cornell Road, Cleveland, Ohio 44106 USA. Tel: 216-368-3670, Fax: 216-368-8964, mark.smithedu