Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Serological markers of HBV...
2. Molecular structures and...
3. How does anti-HBe immunity...
4. Types of HBeAg variants
5. Infection with core promoter...
6. Systems to study the...
7. Core promoter mutations...
8. Some core promoter mutants...
9. Summary and Perspectives
Acknowledgements
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2005; 2(1):2-7. doi:10.7150/ijms.2.2 This issue Cite
Review
Hepatitis B Virus e Antigen Variants
Shuping Tong1, Kyun-Hwan Kim1, Charles Chante2, Jack Wands1, Jisu Li1 ![]()
1. Liver Research Center, Rhode Island Hospital, Brown Medical School, Providence, RI 02903, USA.
2. Cardinal Santos Medical Center, Metro Manila, 1500, Philippines.
Received 2004-10-1; Accepted 2005-1-1; Published 2005-1-5
Abstract
More than 300 million people worldwide are chronically infected with hepatitis B virus (HBV). Considering the very short generation time for a virus, and the high error rate associated with the reverse transcription step of HBV replication, decades of HBV infection are probably equivalent to million years of human evolution. The most important selective force during the natural course of HBV infection appears to be the immune response. The development of anti-HBe antibody in hepatitis B patients usually correlates with reduction of HBV viremia. As a consequence, escape mutants of anti-HBe are selected. The core promoter mutants express less HBe antigen (HBeAg) through transcriptional down regulation, while precore mutants express truncated products. We recently identified additional mutations that modulate HBeAg translation initiation, proteolytic cleavage, and secondary structure maintenance through a disulfide bond. The core promoter mutants have been associated with the development of fulminant hepatitis during acute infection and liver cancer during chronic infection. Consistent with their enhanced pathogenicity, core promoter mutants were found to replicate at up to 10-fold higher levels in transfected human hepatoma cells than the wild-type virus. Moreover, some core promoter mutants are impaired in virion secretion due to missense mutations in the envelope gene. These virological properties may help explain enhanced pathogenicity of core promoter mutants in vivo.
Keywords: Hepatitis B virus, HBeAg, naturally occurring mutations, immune escape, replication, secretion
1. Serological markers of HBV infection
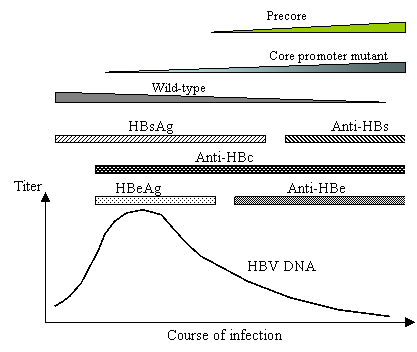
Hepatitis B virus (HBV) chronically infects 300 million people worldwide, and increases their risk to develop hepatocellular carcinoma by a hundred fold [3]. The virus was first discovered as “Australia antigen”, later renamed HBsAg (for hepatitis B surface antigen), in patient blood [6]. HBeAg (hepatitis B e antigen) was identified several years later as a marker for patients at high risk for transmission of the disease [20]. Hepatitis B patients also contain circulating antibodies against HBcAg (hepatitis B core antigen), and will develop antibodies against HBeAg and HBsAg (anti-HBe and anti-HBs) at later stages of infection. Figure 1 depicts the sequential appearance and disappearance of these five serological markers during a typical course of infection. The first stage is characterized by the presence of HBsAg, HBeAg, and IgM class of anti-HBc antibodies, and may last for decades. In the intermediate stage, patients lose HBeAg, develop anti-HBe antibodies, and often enter into clinical remission. Finally, loss of HBsAg and rise of the anti-HBs antibody indicate recovery from infection. With the cloning of the HBV genome, it became apparent that the viremia titer (number of infectious virus particles) is highest during the HBeAg phase of infection, declines by several logs during the anti-HBe phase, and disappears at the anti-HBs phase (Fig. 1).
Disappearance of HBeAg and rise of anti-HBe is associated with decline in viremia titer and replacement of wild-type HBV by the core promoter mutants and/or precore mutants. However, the core promoter mutants become prevalent even before the rise of anti-HBe.
2. Molecular structures and functions of the three viral antigens
Molecular cloning and sequencing of the HBV genome led to the redefinition of the three HBV antigens as viral gene products endowed with specific functions in viral life cycle [for an in-depth review on the molecular biology of HBV, see ref. 13].The HBcAg and HBeAg are alternative translation products of the core gene, with HBeAg translation requiring an upstream precore region ATG codon (Fig. 2).
Expression of core protein and HBeAg. Core protein is translated from pregenomic mRNA, using the ATG codon at 1901 as initiation site. HBeAg is translated from the precore mRNA, using ATG at 1814. The primary translation product is cleaved at the N-terminus by the signal peptidase and in the C-terminus by a basic endopeptidase before secretion into the blood stream. The G1896A nonsense mutation in the precore region specifically prevents translation of HBeAg.

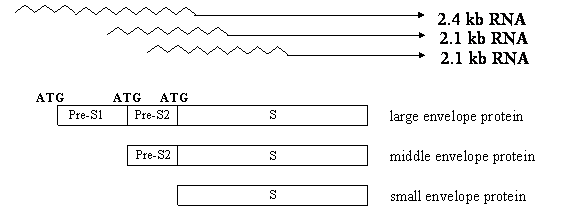
The HBcAg (called “core protein” nowadays) assembles into viral nucleocapsid (core particle), which packages the pregenome (an RNA copy of viral DNA) and polymerase. Inside the core particle, the viral polymerase directs the synthesis of minus strand DNA from the RNA template. It then degrades the RNA pregenome and generates the plus strand DNA via the minus strand template. The HBsAg is the envelope protein of the virus, and actually comprises three co-terminal proteins (large, middle, and small) due to the presence of multiple transcripts and alternative translation initiation sites in the gene (Fig. 3). They contain preS1/preS2/S domains, preS2/S domains, and S domain, respectively. The small envelope protein, composed of S domain alone, is the most abundantly expressed. The envelope proteins interact with the nucleocapsid to initiate its envelopment, and the resultant virus particle (virion) is released into the bloodstream. Thus, HBcAg is not detectable in patient blood unless the envelope is removed. In addition to their incorporation into virus particles, the envelope proteins can be secreted alone as non-infectious subviral particles, which constitute the bulk of HBsAg as detectable in patient blood.
Expression of three co-terminal envelope proteins of the HBV through three in-frame ATG codons and two subgenomic RNA species: 2.4 kb and 2.1 kb transcripts. The 2.4 kb RNA produces the large envelope protein, while the 2.1 kb RNA has heterogeneous 5' ends to allow the expression of middle or small envelope protein. Both large and small envelope proteins have glycosylated and nonglycosylated versions, while the middle protein has monoglycosylated and doubly glycosylated forms.
The N-terminal 29 residues of the HBeAg precursor are specified by the precore region, the first 19 of which serve as the signal peptide to target the protein to the endoplasmic reticulum, where it is cleaved off. Further down the secretory pathway the arginine-rich C-terminus of the molecule is removed, thus releasing mature HBeAg into blood stream (Fig. 2). Therefore, HBeAg differs from core protein by a longer N-terminus and shorter C-terminal tail. However, thanks to an intramolecular disulfide bond HBeAg has a secondary structure quite different from that of core protein [22, 35]. Only one of the two major B cell epitopes of HBeAg is shared with the core protein. HBeAg is not part of the virus particle, and its true function remains not fully understood. Expression of HBeAg is not required for virus replication in vitro [33]. Ablation of e antigen expression had no effect on the in vivo infectivity of the duck hepatitis B virus, but curtailed infection for the woodchuck hepatitis virus (which is more closely related to the human virus) [10, 11, 29]. It was proposed that expression of HBeAg during perinatal infection, the major mode of HBV transmission in Asia, induces immune tolerance. Another potential role of HBeAg in promoting persistent infection is to mimic core protein so as to buffer immune attack of the infected hepatocytes by the anti-HBc antibodies. For a recent review, see ref. [21].
3. How does anti-HBe immunity clear HBV infection?
Of the three antibodies against HBV, anti-HBc develops first, whereas anti-HBs antibody is detected last. The reason for this sequence remains unknown. The HBsAg is the most abundantly expressed protein of HBV, whereas core protein has probably the lowest abundance due to its location inside virus particles. Whether large excess of subviral particles, a unique feature of hepatitis B virus family, delays the development of anti-HBs antibody, has not been experimentally tested. Anti-HBc antibody rises soon after infection but is not associated with change in viremia titer. This could be related to the presence of HBeAg, the variant core protein, as a decoy. The anti-HBe antibody is not expected to directly neutralize viral infectivity, because virus particle does not contain HBeAg. The declined viremia following anti-HBe development could be attributed to loss of HBeAg, which unleashes the anti-viral effect of the anti-HBc immunity. Alternatively, anti-HBe antibodies could destroy infected hepatocytes by recognizing HBeAg on the cell surface, although this aspect remains more or less speculative. The anti-HBs antibodies are known to bind envelope proteins on viral surface to prevent infection. This is the basis for using HBsAg as preventive vaccine against HBV infection.
4. Types of HBeAg variants
The anti-viral effect of anti-HBe immunity may explain the frequent emergence of HBeAg variants in patients with anti-HBe. Since HBeAg expression is not essential for virus replication, the simplest way for the virus to evade the anti-HBe immunity is to switch off HBeAg expression altogether. The so-called “precore mutants” are the first discovered major immune escape mutants of HBV. These mutants are characterized by a G1896A nonsense mutation in the precore region that truncates the precore/core protein into a 28-aa peptide [7, 9, 32]. Other nonsense and frameshift mutations inside the precore region have also been found, although less frequently. Point mutations of the precore ATG codon have also been observed, which prevent initiation of translation. We recently found that triple mutation at the –5, -3, and –2 positions of the precore ATG codon, as occasionally found in some South African strains of HBV, greatly reduced translation efficiency [1]. The selective disruption of HBeAg expression through mutations affecting the precore region rather than the core gene can be easily understood in terms of the indispensable role of core protein for viral replication.
The second common HBeAg variants are the core promoter mutants. They are characterized by point mutations in the promoter for both HBeAg mRNA and core protein mRNA (also called pregenomic RNA) [24]. These mutations were found by transfection experiments to down regulate HBeAg mRNA production, resulting in reduced protein levels [8, 28]. Core promoter mutants are the dominant viral species at not only the anti-HBe stage, but also the late HBeAg stage of infection (Fig. 1). It should be pointed out that the common core promoter mutations, A1762T/G1764A, reduced HBeAg expression by a mere 20% in a genotype A clone that we examined [26]. It is not clear why a moderate reduction in HBeAg expression offers survival advantage, and why the selection is in place well before the rise of anti-HBe.
Considering the many steps required for the secretion of HBeAg, we have recently systemically tested other possible avenues whereby HBeAg expression can be regulated. A V17F missense mutation at the –3 position of the signal peptide cleavage site has been proposed to impair HBeAg production. However, our transfection experiments failed to find a major impact of this mutation on HBeAg secretion (Guarnieri et al., in preparation). On the other hand, naturally occurring mutations at the C-terminal cleavage site were found to reduce HBeAg secretion (Kim et al., in preparation). HBe antigenicity can also be abolished by mutation of one of the two cysteines implicated in the disulfide bond [22, 35]. While mutation of the core gene-derived cysteine into serine or phenylalanine did not interfere with viral replication or virion secretion, substitution of the precore-derived cysteine greatly compromised viral genome replication (Bang et al., Virology, in press). The relevant cysteine codon, when present at the 5' end of pregenomic RNA, constitutes the loop of pregenome encapsidation signal. Finally, we observed an E77Q mutation in the core gene of many naturally occurring core promoter mutants of genotype A (but not from clones with wild-type core promoter sequence), which abolished recognition of both core protein and HBeAg by a rabbit polyclonal antibody (Kim et al., unpublished). This finding is consistent with the localization of the immunodominant epitope of the core protein within residues 77-84 [27, 5]. Whether the selection of this missense mutation is driven by the need to escape the anti-HBc or anti-HBe immunity is an open question.
5. Infection with core promoter mutants is associated with more severe forms of liver diseases
Among the HBeAg variants, the core promoter mutants deserve special attention. Many cases of fulminant hepatitis have been traced to infection with core promoter mutants. However, since fulminant hepatitis is a rare form of acute infection, most reports are descriptive. A case-controlled study is needed to rigorously test whether core promoter mutants are more likely to produce fulminant hepatitis than the wild-type isolates from the same region. Since HBV is considered a noncytocydal virus, the immune response plays a crucial role in generation of liver injury. Therefore, contribution of the genetic makeup of the host in fulminant hepatitis should not be overlooked. It is also necessary to point out that a core promoter mutant often contains many other genetic alterations within the viral genome. Thus, even when a core promoter mutant elicits fulminant hepatitis, mutations elsewhere in the viral genome could be responsible. In one well-documented case, transmission of a core promoter mutant resulted in outbreak of fulminant hepatitis [18]. In another study, a core promoter mutant associated with fulminant hepatitis was found to induce more severe liver damage when experimentally inoculated into chimpanzees [23]. These observations provide compelling evidence for the intrinsic virulence of some core promoter mutants.
During chronic infection, core promoter mutants have been linked to more severe forms of liver diseases including liver cancer. A study from South Africa revealed prevalence of core promoter mutations in 66% of HCC patients but only 11% of asymptomatic carriers matched in age and HBeAg / anti-HBe status [4]. Similarly, core promoter mutations were present in only 3% of Taiwanese inactive carriers but up to 64% of HCC patients [16]. Certainly, prospective epidemiological studies will be needed to demonstrate that rise of core promoter mutations precede cancer development. Another piece of evidence for the enhanced pathogenicity of core promoter mutants came from comparative studies of HBV genotypes. East Asian patients are primarily infected with genotype C or B of HBV, with a North to South transition. Interestingly, genotype C patients often suffer from more severe liver diseases, delayed HBeAg to anti-HBe seroconversion, and accelerated HCC development as compared with genotype B patients [reviewed in ref. 12]. Further analysis revealed that genotype C isolates are more likely to develop core promoter mutations than genotype B [16, 25, 30]. It has been recently suggested that core promoter mutations, rather than genotype C per se, are the primary risk factor for liver cancer [37]. Like core promoter mutations, the G1896A HBeAg-negative precore mutation develops late in the course of HBV infection. However, the prevalence of the precore mutation was not elevated in cancer patients relative to matched controls [37]. Thus, the association between core promoter mutations and liver cancer is genuine.
6. Systems to study the biological properties of HBV variants
Although molecular epidemiological surveys have provided circumstantial evidence for the increased pathogenicity of core promoter mutants, observations in patients are complicated by variables such as individual differences in susceptibility to virus infection or replication, the vigor of the immune response, and coexistence of viral quasispecies. Experimental approaches are required to validate the association between each viral genotype and certain biological / pathobiological phenotypes, and to map the responsible mutation within the genome. Unfortunately, due to the strict host specificity of hepatitis B virus only chimpanzees are susceptible to experimental infection. Moreover, chimpanzees typically do not reproduce the liver damage seen with human infection. In this regard, tupaias are susceptible to transient infection with HBV [34]. An alternative in vivo model is woodchuck hepatitis virus, which shares about 80% of sequence homology with HBV. However, it is not certain whether mutations introduced into the homologous position in the woodchuck hepatitis virus genome will reproduce the same biological effects. A simple system to study the biological (but not pathobiological) properties of HBV variants is human hepatoma cell lines such as Huh7 and HepG2. When transfected with certain forms of cloned HBV genomes (such as tandem dimer), these cells support one round of HBV gene transcription, protein translation, genome replication, and virion formation / secretion. When coupled with current techniques in the amplification of the full-length HBV genome from patient blood, cloning, mutagenesis, and efficient transient transfection reagents, this system provides a powerful tool to study the regulation of viral gene expression, genome replication, and virion assembly. Certainly, human hepatoma cells are different from primary human hepatocytes. In this regard, a differentiated human hepatocyte cell line has been established. Under appropriate conditions, this cell line can be rendered susceptible to HBV infection [14].
7. Core promoter mutations cumulatively enhance viral genome replication in vitro
While a single nucleotide change or insertion/deletion is often present in precore mutants and sufficient to abolish HBeAg expression, the number and position of mutations in the core promoter vary. The mutations are clustered around nucleotides 1750—1770, with the A1762T and G1764A being the most common. Therefore, the A1762T/G1764A double mutation has been chosen for further characterization through site-directed mutagenesis and transfection experiments. Many independent studies have been performed and the double mutation appears to reduce HBeAg expression (by only 20% in our hand) and double the genome replication capacity [8, 28, 31, 36]. From a different perspective, the HBV isolate implicated in fulminant hepatitis outbreak was found to replicate at least 10 times higher levels than a wild-type clone [15]. This genome is both a core promoter mutant and precore mutant. It contained A1762T, G1764A, C1766T, and T1768A mutations in the core promoter. Mapping experiments and site-directed mutagenesis revealed the 1766/1768 double mutation, rather than common mutations at 1762 and 1762, as responsible for the enhancement of viral replication [2].
In collaboration with Dr. Zoulim from France, we recently searched for naturally occurring HBV genomes with high replication capacity. We chose HBeAg positive patients so as to avoid the effect of immune response on virus titers. Patients infected with genotype A of HBV, with either very high or very low viremia titers, were studied. Surprisingly, clones derived from highly viremic patients uniformly displayed low replication capacity, while some clones derived from low viremia patients had much higher replication levels [26]. Sequence analysis revealed core promoter mutations in the high replicating but not low replicating clones. The highest replicating clones contained T1753C/A1762T/G1764A/C1766T quadruple mutation or 1762/1764/1766 triple mutation, and the next highest replicating clone contained 1753/1762/1764 mutations. Site-directed mutagenesis of a wild-type clone revealed 2-, 4-, 8-, and 8- fold enhancement of viral replication by the 1762/1764,1753/1762/1764, 1762/1764/1766, and 1753/1762/1764/1766 mutations, respectively (Table 1) [26]. These mutations reduced HBeAg expression by 20%, 30%, 75%, and 80%, respectively. These results provide compelling evidence that core promoter mutations enhance viral genome replication and reduce HBeAg expression in a cumulative manner. In this regard, the 1762/1764 mutations emerge first, followed by the less common mutations in the core promoter. Our findings suggest the gradual loss of HBeAg expression and enhancement of viral replication capacity over the course of chronic HBV infection.
Cumulative effect of core promoter mutations on viral genome replication and HBeAg expression
| Core promoter mutations | Genome replication (fold) | HBeAg expression (level) |
| None | 1 | 100% |
| 1762/1764 | 2 | 80% |
| 1753/1762/1764 | 4 | 70% |
| 1762/1764/1766 | 8 | 25% |
| 1753/1762/1764/1766 | 8 | 20% |
8. Some core promoter mutants are impaired in virion secretion due to mutated envelope gene
Clones 4B and 4C are derived from the same patient and both displayed extremely high replication capacity due to the 1753/1762/1764/1766 quadruple mutation in the core promoter region [26]. However, clone 4B secreted virus particles to culture medium very efficiently, while clone 4C was totally defective in virion secretion. It also failed to secrete HBsAg into culture supernatant despite its presence in cell lysate (Table 2). Another high replicating core promoter mutant, clone 3.4, was impaired in virion secretion and displayed low HBsAg levels in both the cell lysate and culture supernatant [26]. Extensive mapping experiments revealed an R169P mutation in the S gene of clone 4C as responsible for the block to the secretion of both viral and subviral particles [17]. For clone 3.4, a G119E mutation in the S gene impaired virion secretion. This mutation apparently also impaired HBsAg recognition by the monoclonal antibody used for the commercial assay, since residue 119 is in the vicinity of the determinant, the dominant epitope in the S domain. Clone 4B actually contained a mutation (I110M) capable of blocking virion secretion. However, presence of an M133T mutation in this clone overrides the I110M mutation and confers efficient virion secretion [17]. Interestingly, the M133T mutation creates a consensus sequence for N-linked glycosylation (NST), which may facilitate proper protein folding or assembly through the disulfide bonds.
Effect of mutations in the envelope gene on secretion of viral and subviral particles as well as HBs antigenicity
| clone | nt change | S domain Env proteins | RT domain polymerase | Effect |
| 3.4 | G510A | G119E | silent | reduces both intra- and extra-cellular HBsAg; impairs virion secretion |
| 4B | T484G | I110M | S466A | blocks virion secretion |
| 4B | T552C | M133T | silent | suppresses G510A and T484G mutations; creates an N glycosylation site? |
| 4C | G660C | R169P | silent | abolishes HBsAg secretion; abolishes virion secretion |
9. Summary and Perspectives
The development of anti-HBe immunity can be regarded as a turning point in the battle between HBV and its host. Whatever the mechanism, the anti-HBe antibody helps control viral replication to a much reduced level. Certainly, complete clearance of HBV replication would await the rise of anti-HBs antibody, which prevents spread of virus infection. The unusual observation that some Mediterranean patients maintain high viremia titers despite the presence of anti-HBe antibody provided impetus for studies that led to the discovery of the precore mutants with defective HBeAg expression [9, 32]. It is now clear that emergence of G1896A precore mutants is a common feature for most viral genotypes, since it improves rather than disrupts a base pair in the pregenome encapsidation signal [19]. However, in most patients the viremia titer declines markedly despite the emergence of such “immune escape mutants”. The reason for the reduced viral replication in most patients despite precore mutation remains unknown.
Although the core promoter mutants were initially identified as HBeAg variants, they have been associated with fulminant hepatitis and liver cancer in vivo and found to display enhanced replication and sometimes impaired virion secretion in vitro. It is tempting to suggest that the enhanced replication capacity and reduced virion secretion may increase viral load in the liver, thus triggering liver damage either directly or indirectly through the immune response. When massive liver damage occurs during acute infection, fulminant hepatitis may ensue. When such damage occurs during chronic infection, it increases hepatocyte turnover, induces fibrosis, and enhances the chance of hepatocellular transformation and malignancy. This hypothesis is best examined in an in vivo model, such as HBV transgenic mice, although we don't know whether the high replication phenotype of core promoter mutants will be preserved in the mouse liver. We also need to characterize more naturally occurring core promoter mutants to determine whether impaired virion secretion is a common feature of such mutants.
Acknowledgements
We thank Dr. Zoulim, Lyon, France, for collaboration on this project and many helpful discussions. This work was supported by grants AI54535, DK62857, and p20RR15578 from the National Institutes of Health, and by the Tan Yan Kee Foundation.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Ahn S, Kramvis A, Kawai S, Spangenberg H, Li J, Kimbi G, Kew M, Wands J, Tong S. Sequence variation upstream of precore translation initiation codon reduces hepatitis B virus e antigen production. Gastroenterology. 2003 ;125:1370-1378
2. Baumert T, Rogers S, Hasegawa K, Liang T. Two core promoter mutations identified in a hepatitis B virus strain associated with fulminant hepatitis result in enhanced viral replication. J. Clin. Invest. 1996 ;98:2268-2276
3. Beasley R, Hwang L, Lin C, Chien C. Hepatocellular carcinoma and hepatitis B virus: A prospective study of 22707 men in Taiwan. Lancet. 1981 ;2:1129-1133
4. Baptista M, Kramvis A, Kew M. High prevalence of 1762T 1764A mutations in the basic core promoter of hepatitis B virus isolated from Black Africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology. 1999 ;29:946-953
5. Belnap D, Watts N, Conway J, Cheng N, Stahl S, Wingfield P, Steven A. Diversity of core antigen epitopes of hepatitis B virus. Proc. Natl. Acad. Sci. USA. 2003 ;100:10884-10889
6. Blumberg B, Alter H, Visnich S. A “new” antigen in leukemia sera. JAMA. 1965 ;191:541-546
7. Brunetto MR, Stemler M, Schodel F, Will H, Ottoberlli A, Rizzetto M, Bonino F. Identification of HBV variants which cannot produce precore derived HBeAg and may be responsible for severe hepatitis. Ital. J. Gastroenterol. 1989 ;21:151-154
8. Buckwold V, Xu Z, Chen M, Yen T, Ou J. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J. Virol. 1996 ;70:5845-5851
9. Carman W, Jacyna M, Hadziyannis S, Karayiannis P, McGarvey M, Makris A, Thomas H. Mutation preventing formation of the hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989 ;2:588-591
10. Chang C, Enders G, Sprengel R, Peters N, Varmus H, Ganem D. Expression of the precore region of an avian hepatitis B virus is not required for viral replication. J. Virol. 1987 ;61:3322-3325
11. Chen H, Kew M, Hornbuckle W, Tennant B, Cote P, Gerin J, Purcell R, Miller R. The precore gene of the woodchuck hepatitis virus genome is not essential for viral replication in the natural host. J. Virol. 1992 ;66:5682- 5684
12. Chu C, Lok A. Clinical significance of hepatitis B virus genotypes. Hepatology. 2002 ;35:1274-1276
13. Ganem D, Schneider R. Hepadnaviridae: the viruses and their replication. Knipe D, Howley P, eds. : Fields Virology. 2001:2923-2970
14. Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA. 2002 ;99:15655-15660
15. Hasegawa K, Huang J, Rogers S, Blum H, Liang T. Enhanced replication of a hepatitis B virus mutant associated with an epidemic of fulminant hepatitis. J. Virol. 1994 ;68:1651-1659
16. Kao J, Chen P, Lai M, Chen D. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003 ;124:327-334
17. Khan N, Guarnieri M, Ahn S, Li J, Zhou Y, Bang G, Kim K, Wands J, Tong S. Modulation of hepatitis B virus secretion by naturally occurring mutations in the S gene. J. Virol. 2004 ;78:3262-3270
18. Liang T, Hasegawa K, Rimon N, Wands J, Ben-Porath E. A hepatitis B virus mutant associated with an epidemic of fulminant hepatitis B. New Engl. J. Med. 1991 ;324:1705-1709
19. Li J, Tong S, Wen Y, Vitvitski L, Zhang Q, Trepo C. Hepatitis B virus genotype A rarely circulates as an HBe-minus mutant: possible contribution of a single nucleotide in the precore region. J. Virol. 1993 ;67:5402-5410
20. Magnius L, Espmark J. New specificities in Australia antigen positive sera distinct from the Le Bouvier determinants. J. Immunol. 1972 ;109:1017-1021
21. Milich D, Liang J. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology. 2003 ;38:1075-1086
22. Nassal M, Rieger A. An intramolecular disulfide bridge between Cys-7 and Cys61 determines the structure of the secretory core gene product (e antigen) of hepatitis B virus. J Virol. 1993 ;67:4307-4315
23. Ogata N, Miller R, Ishak K, Purcell R. The complete nucleotide sequence of a pre-core mutant of hepatitis B virus implicated in fulminant hepatitis and its biological characterization in chimpanzees. Virology. 1993 ;194:263-276
24. Okamoto H, Tsuda F, Akahane Y, Sugai Y, Yoshiba M, Moriyama K, Tanaka T, Miyakawa Y, Mayumi M. Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J. Virol. 1994 ;68:8102-8110
25. Orito E, Mizokami M, Sakugawa H, Michitaka K. et al. A case-control study for clinical and molecular biological differences between hepatitis B viruses of genotypes B and C. Hepatology. 2001 ;33:218-233
26. Parekh S, Zoulim F, Ahn S, Tsai A, Li J, Kawai S, Khan N, Trepo C, Wands J, Tong S. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J. Virol. 2003 ;77:6601-6612
27. Salfeld J, Pfaff E, Noah M, Schaller H. Antigenic determinants and functional domains in core antigen and e antigen from hepatitis B virus. J. Virol. 1989 ;63:798-808
28. Scaglioni P, Melegari M, Wands J. Biological properties of hepatitis B viral genomes with mutations in the precore promoter and precore open reading frame. Virology. 1997 ;233:374-381
29. Schlicht H, Salfeld J, Schaller H. The duck hepatitis B virus pre-C region encodes a signal sequence which is essential for synthesis and secretion of processed core proteins but not for virus formation. J. Virol. 1987 ;61:3701-3709
30. Sumi H, Yokosuka O, Seki N, Arai M. et al. Influence of hepatitis B virus genotypes on the progression of chronic type B liver disease. Hepatology. 2003 ;37:19-26
31. Tang H, Raney A, McLachlan A. Replication of the wild type and a natural hepatitis B virus nucleocapsid promoter variant is differentially regulated by nuclear hormone receptors in cell culture. J. Virol. 2001 ;75:8937- 8948
32. Tong S, Li J, Vitvitski L, Trepo C. Active hepatitis B virus replication in the presence of anti-HBe is associated with viral variants containing an inactive pre-C region. Virology. 1990 ;176:596-603
33. Tong S, Diot C, Gripon P, Vitvitski L, Trepo C, Guguen-Guillouzo C. In vitro replication competence of a cloned hepatitis B virus variant with a nonsense mutation in the distal pre-C region. Virology. 1991 ;181:733-737
34. Walter E, Keist R, Niederost B, Pult I, Blum HE. Hepatitis B virus infection of tupaia hepatocytes in vitro and in vivo. Hepatology. 1996 ;24:1-5
35. Wasenauer G, Kock J, Schlicht H. Relevance of cysteine residues for biosynthesis and antigenicity of human hepatitis B virus e protein. J Virol. 1993 ;67:1315-1321
36. Yu X, Mertz J. Distinct modes of regulation of transcription of hepatitis B virus by the nuclear receptors HNF4 and COUP-TF1. J. Virol. 2003 ;77:2489-2499
37. Yuen M, Tanaka Y, Mizokami M, Yuen C, Wong D. et al. Role of hepatitis B virus genotypes Ba and C, core promoter and precore mutations of hepatocellular carcinoma: a case control study. Carcinogenesis. 2004 ;25:1593-1598
Author biography
Shuping Tong MD, PhD is an Assistant Professor of Medicine at the Liver Research Center, Rhode Island Hospital, Brown Medical School. Dr. Tong was among the first to independently identify the precore mutants of HBV, for which he received the "Young Investigator's Award" from the French Society for the Study of Liver Diseases in 1989. His major research interests are on the molecular properties of naturally occurring HBV variants, with regard to gene expression, genome replication, and virion secretion.
Kyun-Hwan Kim PhD is a Research Fellow at the Liver Research Center, Rhode Island Hospital, Brown Medical School. He works on the mutations in the HBV genome that modulate core protein and e antigen expression.
Charles Chante MD is Professor of Medicine, Medical Director of the Cardinal Santos Medical Center and Chief of the Gastroenterology Division. Dr. Chante has broad interest and experience in treating diseases of the Gastrointestinal tract, including chronic hepatitis B and liver cancer.
Jack Wands MD is Professor of Medicine, Director of the Liver Research Center, Rhode Island Hospital, Brown Medical School, and Chairman of the Division of Gastroenterology at Brown Medical School. His research interests include hepatitis B and C viruses, liver cancer, signal transduction in the liver, gene therapy, and effects of chronic ethanol exposure.
Jisu Li MD, PhD is an Assistant Professor of Medicine at the Liver Research Center, Rhode Island Hospital, Brown Medical School. Dr. Li was the first to demonstrate the biological and clinical significance of HBV genotypes when she discovered rare emergence of precore mutants in genotype A strains due to a base-pairing requirement of the overlapping pregenome encapsidation signal. She is interested in studies on the molecular biology of HCV, the duck hepatitis B virus receptor complex, and HBV genotypes.
![]() Corresponding address:
Corresponding address:
Shuping Tong or Jisu Li, The Liver Research Center, Rhode Island Hospital and Brown Medical School, 55 Claverick Street, Providence, RI 02906. Telephone: 401-444-7365 (ST); 401-444-7387 (JL). Fax: 401-444-2939. E-mail: Shuping_Tong_MDedu; Ji_Su_Li_MDedu