Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Biochemical mechanism of...
3. Classification
4. Oncogenic activation of...
5. Tyrosine kinases as targets...
6. Mining the kinome to meet the...
7. Conclusion
Abbreviations
References
Tables and Figures
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2004; 1(2):101-115. doi:10.7150/ijms.1.101 This issue Cite
Review
Tyrosine kinase – Role and significance in Cancer
Manash K. Paul, Anup K. Mukhopadhyay ![]()
Department of Biotechnology, National Institute of Pharmaceutical Education and Research, Sector-67, S.A.S Nagar, Mohali, Punjab, India-160062
Received 2004-3-30; Accepted 2004-5-15; Published 2004-6-1
Abstract
Tyrosine kinases are important mediators of the signaling cascade, determining key roles in diverse biological processes like growth, differentiation, metabolism and apoptosis in response to external and internal stimuli. Recent advances have implicated the role of tyrosine kinases in the pathophysiology of cancer. Though their activity is tightly regulated in normal cells, they may acquire transforming functions due to mutation(s), overexpression and autocrine paracrine stimulation, leading to malignancy. Constitutive oncogenic activation in cancer cells can be blocked by selective tyrosine kinase inhibitors and thus considered as a promising approach for innovative genome based therapeutics. The modes of oncogenic activation and the different approaches for tyrosine kinase inhibition, like small molecule inhibitors, monoclonal antibodies, heat shock proteins, immunoconjugates, antisense and peptide drugs are reviewed in light of the important molecules. As angiogenesis is a major event in cancer growth and proliferation, tyrosine kinase inhibitors as a target for anti-angiogenesis can be aptly applied as a new mode of cancer therapy. The review concludes with a discussion on the application of modern techniques and knowledge of the kinome as means to gear up the tyrosine kinase drug discovery process.
Keywords: Tyrosine kinase, Cancer, Oncogenic activation, Inhibitor, Drug discovery
1. Introduction
Multicellular organisms live in a complex milieu where signaling pathways contribute to critical links, for their existence. Tyrosine kinases are important mediators of this signal transduction process, leading to cell proliferation, differentiation, migration, metabolism and programmed cell death. Tyrosine kinases are a family of enzymes, which catalyzes phosphorylation of select tyrosine residues in target proteins, using ATP. This covalent post-translational modification is a pivotal component of normal cellular communication and maintenance of homeostasis [1,2] . Tyrosine kinases are implicated in several steps of neoplastic development and progression. Tyrosine kinase signaling pathways normally prevent deregulated proliferation or contribute to sensitivity towards apoptotic stimuli. These signaling pathways are often genetically or epigenetically altered in cancer cells to impart a selection advantage to the cancer cells. Thus, it is no wonder that aberrant enhanced signaling emanating from tyrosine kinase endows these enzymes a dominating oncoprotein status, resulting in the malfunctioning of signaling network [3] .
The discovery that SRC oncogene having a transforming non receptor tyrosine kinase activity [4] , and the finding of EGFR, the first receptor tyrosine kinase paved the way to the understanding of the role and significance of tyrosine kinase in cancer [5] . With the deciphering of the Human Genome Project more than 90 tyrosine kinases have been found out. The more science entangles the intricacies of cellular signaling the more we find the involvement of tyrosine kinase in cellular signaling circuits that are implicated in cancer development. Tyrosine kinases represent a major portion of all oncoprotein that play a transforming role in a plethora of cancers. Hence the identification and development of therapeutic agents for disease states that are linked to abnormal activation of tyrosine kinases due to enhanced expression, mutation or autocrine stimulation leading to abnormal downstream oncogenic signaling have taken a centre stage as a potent target for cancer therapy [6,7] .
2. Biochemical mechanism of action of tyrosine kinase
Tyrosine kinases are enzymes that selectively phosphorylates tyrosine residue in different substrates. Receptor tyrosine kinases are activated by ligand binding to their extracellular domain. Ligands are extracellular signal molecules (e.g. EGF, PDGF etc) that induce receptor dimerization (except Insulin receptor). Different ligands employ different strategies by which they achieve the stable dimeric conformation. One ligand may bind with two receptor molecules to form 1:2 ligand: receptor complex e.g. growth hormone and growth hormone receptor, while in other cases two ligands binds simultaneously to two receptors 2:2 ligand receptor complex and provides the simplest mechanism of receptor dimerization e.g. VEGF and VEGFR. The receptor dimerization is also stabilized by receptor–receptor interactions. Some ligand receptor is not sufficient for some complex and is stabilized by accessory molecules e.g. FGFs are unable to activate FGFR complex and is stabilized by heparin sulfate proteoglycans (HSPG). Ligand binding to the extracellular domain stabilizes the formation of active dimmers and consequently protein tyrosine kinase activation.
Structural studies of the catalytic core of several RTKs, supported by biochemical and kinetic studies of receptor phosphorylation have provided proof that receptor oligomerization increases the local concentration of the RTKs, leading to efficient transphosphorylation of tyrosine residues in the activation loop of the catalytic domain. Upon tyrosine phosphorylation the activation loop adopts an open conformation that gives access to ATP and substrates and makes ATP transfer from Mg-ATP to tyrosine residue on the receptor itself and on cellular proteins involved in signal transduction.
The ATP binding intracellular catalytic domain that catalyzes receptor autophosphorylation displays the highest level of conservation between the RTKs. The ATP binding site serves as a docking site for specific binding of cytoplasmic signaling proteins containing Src homology-2 (SH2) and protein tyrosine binding (PTB) domains. These proteins in turn recruit additional effector molecules having SH2, SH3, PTB and Pleckstrin homology (PH) domain. This results in the assembly of signaling complexes to the activated receptor and the membrane and subsequent activation of a cascade of intracellular biochemical signals, which leads to the activation or repression of various subsets of genes and thus defines the biological response to signals. During these processes, receptors migrate within the plasma membrane and are internalized through clathrin-coated invagination, which eventually seal off and forms an endocytic vesicle. The endocytic vesicles fuse with the lysosomes and in the process the receptor and ligand may be degraded by the lysosomal enzymes. The receptors are also recycled in some cases. During the whole process of receptor internalization the ligand receptor complex is dissociated and this results in the termination of the signaling reaction.
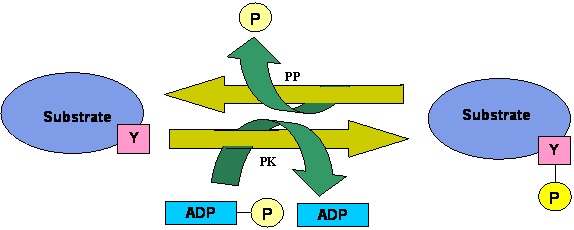
Schematic representation of the mode of action of tyrosine kinase. PK represents protein kinase and PP stands for protein phosphatase.
3. Classification
Tyrosine kinases are primarily classified as receptor tyrosine kinase (RTK) e.g. EGFR, PDGFR, FGFR and the IR and non-receptor tyrosine kinase (NRTK) e.g. SRC, ABL, FAK and Janus kinase. The receptor tyrosine kinases are not only cell surface transmembrane receptors, but are also enzymes having kinase activity. The structural organization of the receptor tyrosine kinase exhibits a multidomain extracellular ligand for conveying ligand specificity, a single pass transmembrane hydrophobic helix and a cytoplasmic portion containing a tyrosine kinase domain. The kinase domain has regulatory sequence both on the N and C terminal end [2,8] .
NRTK are cytoplasmic proteins, exhibiting considerable structural variability. The NRTK have a kinase domain and often possess several additional signaling or protein-protein interacting domains such as SH2, SH3 and the PH domain [9] . The tyrosine kinase domain spans approximately 300 residues and consists of an N terminal lobe comprising of a 5 stranded β sheet and one α helix, while the C terminal domain is a large cytoplasmic domain that is mainly α helical. ATP binds in the cleft in between the two lobes and the tyrosine containing sequence of the protein substrate interacts with the residues of the C terminal lobe. RTK are activated by ligand binding to the extracellular domain followed by dimerization of receptors, facilitating trans-phosphorylation in the cytoplasmic domain whereas the activation mechanism of NRTK is more complex, involving heterologous protein-protein interaction to enable transphosphorylation [10] .
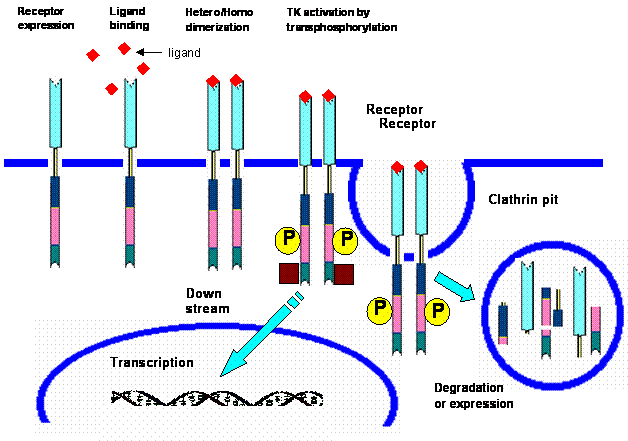
Mechanism of action of tyrosine kinase. 1. Receptor expression at membrane claveola 2. Ligand binding 3. Hetero/homodimerization leading to tyrosine kinase activation and tyrosine transphosphorylation 4. Signal transduction 5. Receptor internalization 6. Receptor degradation or re-expression.
4. Oncogenic activation of tyrosine kinase
Normally the level of cellular tyrosine kinase phosphorylation is tightly controlled by the antagonizing effect of tyrosine kinase and tyrosine phosphatases. There are several mechanisms by which tyrosine kinase might acquire transforming functions, but the ultimate result is the constitutive activation of normally controlled pathways leading to the activation of other signaling proteins and secondary messengers which serves to hamper the regulatory functions in cellular responses like cell division, growth and cell death [11] . Constitutive activation of tyrosine kinase may occur by several mechanisms.
4.1. Activation by mutation
An important mechanism leading to tyrosine kinase deregulation is mutation. Mutations within the extracellular domain e.g. EGFRv III mutant lacks amino acid 6-273 which gives rise to receptor tyrosine kinase constitutive activity, that leads to cell proliferation in the absence of ligand in glioblastomas, ovarian tumors and non small cell lung carcinoma [12] . Point mutations either in the extracellular domain of the FGFR 3 results in an unpaired cysteine residue allowing abnormal receptor dimerization through intermolecular disulfide bonding, or in the activation loop of kinase domain were identified in multiple myeloma. Somatic mutations in the EGFR 2 and 3 have been associated with human bladder and cervical carcinomas. Breakpoints of abnormal chromosomal translocation are also an important source of mutation [13] .
4.1.1. BCR-ABL and human leukemia
CML is a chronic myelodysplastic hematopoietic stem cell disorder syndrome (95% of the CML) resulting from a reciprocal translocation between chromosome-9 and chromosome-22, the Philadelphia chromosome. Break point cluster region (BCR) sequences of chromosome-22 on translocation juxtaposes with the c-ABL tyrosine kinase of chromosome-9. The fusion gene produces a 210 KDa mutant protein in which the first exon of c-ABL has been replaced by BCR sequences, encoding either 927 or 902 amino acid. Another BCR-ABL fusion protein of 185 KDa containing BCR sequences from exon 1fused to exon 2-11 of c-ABL, is found in 10% of adult ALL patients. The BCR-ABL chimeric gene product has a tyrosine kinase activity several fold higher than it's normal counterpart and correlates with the disease phenotype [14,15,16,17,18] .
4.1.2. TEL-ABL and human leukemia
TEL-ABL tyrosine kinase like ABL-BCR is constitutively phosphorylated due to reciprocal translocation t(9,12) in case of ALL and with a complex karyotype t(9,12,14) in patients with CML. TEL which is a putative transcription factor is fused in-frame with exon-2 of the ABL proto-oncogene, producing a fusion protein product with elevated tyrosine kinase activity [14,19] . Other important translocations include t(5,12) in CMML producing TEL-PDGF receptor. The helix-loop-helix motif of TEL is believed to induce homodimerization and kinase activation of the TEL-ABL and TEL-PDGFRβ fusion proteins. NPM-ALK fusion products t(2,5) is constitutively activated in Anaplastic large cell lymphoma [20,21] .
4.2. Autocrine-Paracrine loops
Autocrine-paracrine stimulation serves as an important mechanism for the constitutive activation of tyrosine kinase specially receptor tyrosine kinases. This activation loop is stimulated when a receptor tyrosine kinase is abnormally expressed or overexpressed in presence of it's associated ligand or when there is an overexpression of the ligand in presence of it's cognate receptor. A role of autocrine-paracrine stimulation has been immanent in a variety of human cancers. Burning examples are being provided by EGFR, PDGFR and IGF receptors and their associated ligands [22] .
4.2.1. EGFR in autocrine paracrine loops
Strong associations have been studied among the expression of EGFR and it's primary ligand EGF and TGFα in many human cancers including non small cell lung cancer [22] , bladder cancer, breast cancer and glioblastoma multiforme. Increased expression of EGFR is reported in 40-80% of non small cell lung cancer and is also overexpressed in 50% of primary lung cancer [15] . TGFα another ligand of EGFR is also involved in autocrine paracrine growth loops in 20-40% of lung cancer [23] . EGFR overexpression of wild type or mutated forms is documented in 40% of cases of glioblastoma multiforme [24] and 40%-50 % of bladder cancer patient [25] .
4.2.2. PDGFR in Autocrine paracrine Loop
Concurrent expression of PDGFR and it's cognate ligand PDGF-A and PDGF-B has been observed in most astrocytic brain tumors and gliomas [26] .
4.2.3. Insulin like growth factor receptors in Autocrine growth loop
Coexpression of IGFR and its ligand IGF I and IGF II is reported in the pathogenesis of breast cancer, prostrate cancer and small cell lung cancer [27] . Elevated IGF-I R autophosphorylation and kinase activity has been reported in breast cancer [28] . Elevated prostrate cancer risk is also correlated with elevated plasma IGF-I levels [29] . Hence a strong association of autocrine paracrine loops has been implicated in the pathogenesis of several cancers.
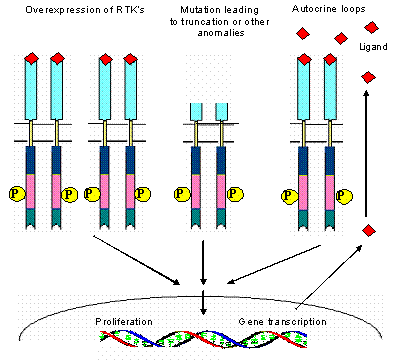
Schematic view of different mechanisms leading to the constitutive activation of tyrosine kinase.
5. Tyrosine kinases as targets for anticancer agents
The role of tyrosine kinases in cancer molecular pathogenesis is immense and recently kinases have come in vogue as potential anticancer drug targets, as a result a couple of anticancer drugs are in the market. The complexity and the number of tyrosine kinases have greatly increased with the sequencing effort of the Human Genome Project, thus providing more opportunities for drug discovery. Recent understanding of the molecular pathophysiology of cancer have highlighted that many tyrosine kinases are found upstream or downstream of epidemiologically relevant oncogenes or tumor suppressor, in particular the receptor tyrosine kinases [30] .
5.1. Site of targeting
Cancer research got a boom with the declaration by the US president Richard Nixon in The National Cancer Act (1971). By the late 1980 there were evidence of low molecular weight tyrosine kinase inhibitors. Inhibiting the activity of tyrosine kinases by low molecular weight compounds capable of interfering with either ligand binding (in the case of receptor tyrosine kinases) [31] or with protein substrate (in case of non receptor tyrosine kinase) has proved to be difficult [32] . Although the bisubstrate inhibitor approach offered promise, but with very little practical progress. Approaches to generate non-competitive or allosteric inhibitors have also failed. The ATP competitive inhibitors appear to be the target of choice [33] .
5.2. The ATP binding site
ATP binds within a deep cleft formed between the two lobes of the tyrosine kinase domain. Though the ATP binding site is highly conserved the architecture in the regions proximal to the ATP binding site does afford some key diversity for designing new drug and has potential application in drug discovery.
The ATP binding sites have the following features-
a) Adenine region – contains two key Hydrogen bonds formed by the interaction of N-1 and N-6 amino group of the adenine ring. Many potent inhibitors use one of these Hydrogen bonds.
b) Sugar region – a hydrophilic region, except a few e.g. EGFR. Hydrophobic pocket – though not used by ATP but plays an important role in inhibitor selectivity.
c) Hydrophobic channel – not used by ATP and may be exploited for inhibitor specificity.
d) Phosphate binding region – can be used for improving inhibitor selectivity [34].
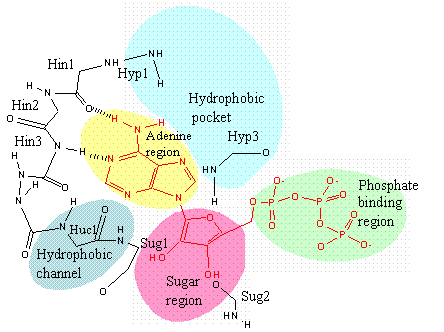
Model of the ATP-binding site of protein kinases. ATP is depicted in red, Sug1, Hyp1 & Hyc1 are residues lining the sugar region, hydrophobic pocket (Hyp), hydrophobic channel (Huc) and hinge region (Hin) respectively (redrawn) [34].
5.3. Small molecule inhibitor
Tyrosine kinase forms a significant share of all oncoproteins thus they take centre stage as possible targets for cancer therapy. Hence low molecular weight tyrosine phosphorylation inhibitors (tyrphostins) have been proposed to be prospective anti-proliferating agents. By late 1980s it was proved that low molecular weight EGFR inhibitors could block EGF dependent cell proliferation [35] . Within a short time reports concerning effective tyrosine kinase inhibitors was the key promising development in anticancer development. With further research in the following years it was quite clear that neglected kinase targets against the biases have come into vogue [36] . Most of the tyrosine kinase inhibitors generated since have been ATP mimics. Interestingly many tyrphostins that possess one aromatic ring are substrate mimics [37] . These substrate mimics can be interestingly converted into ATP mimics once the nitrogen of the characteristic benzene malonitrile is incorporated into the second ring. Compounds that are found to be ATP mimics possess at least two aromatic rings. The ATP binding site though evolutionarily conserved can be selectively targeted by taking advantage of the minor difference in the kinase domain. The minor difference leads to changes in hydrogen bonding and hydrophobic interactions resulting in differences of affinity [38] .
Of all the tyrosine kinase inhibitors the most successful are Gleevec, Iressa and Tarceva. The novel anticancer drug Gleevec/ Glivec/ Imatinib mesylate (Novartis STI571) is a success for CML and c-kit positive metastatic GIST. Gleevec selectively and effectively inhibits the kinase activity of BCR-ABL fusion protein, which is responsible for the constitutive proliferative signaling. It also inhibits TEL-ABL and TEL-PDGFR fusion proteins. STI571 remains bound to the ATP binding cleft of the unphospholrylated (activation loop) Abl, thus establishing extensive contacts with residues lining the cleft and with peptide segments just outside the cleft. A large change in conformation of the nucleotide binding domain is accompanied with the binding of the drug. The binding of STI571 prevents ATP to access the ATP binding cleft and thus inhibits subsequent tyrosine phosphorylation of the substrate [34,36,39] . Iressa is a selective inhibitor of EGF receptor tyrosine kinase in non small cell lung cancer and squamous cell carcinoma [40] .
The downstream target of PI3K pathway, mTOR (non receptor tyrosine kinase) responsible for abnormal cellular growth and proliferation in glioblastoma and renal cancer is inhibited by rapamycin and CCI779. These two compounds are currently in phase II studies [41] .
Structure of some of the important protein kinase inhibitors.
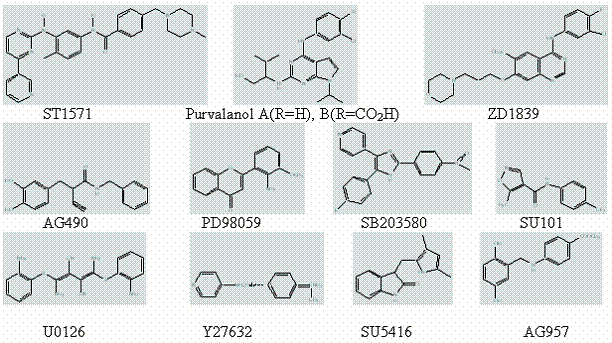
5.4. Monoclonal antibody
The extracellular domain of the receptor tyrosine kinase provides an excellent target for monoclonal antibodies. With the advancement of genomics, design, selection and production of therapeutic monoclonal antibodies has become much more easier. The revolution in antibody technology now allows us to produce humanized, human chimeric or bispecific antibody for targeted cancer therapy [31,42] .
The EGFR family of receptor tyrosine kinase comprises of four members – the EGFR/ Erb B1, HER-2/ Erb B2, HER-3/ Erb B3 and HER-4/ Erb B4. Members of the EGFR family are involved in some complex biological signal transduction network. Both EGFR and HER-2/ neu are amplified in tumor samples of breast, lung and colorectal tissues. Their overexpression leads to elevated MAP kinase and PI3 kinase recruitment and subsequent suppression of apoptosis, deregulation of cell cycle and proliferation.
Based on the discovery of the importance of HER-2 gene amplification in breast cancer Herceptin (Transtuzumab) was developed by Genetech in 1988 and is one of the best examples of targetted next generation anticancer therapeutic for cancer. Tyrosine kinases are key regulators of cell survival and proliferation in breast cancer [42,43] . The orphan receptor tyrosine kinase HER2 gene is amplified in breast cancer and acts as a major signaling partner for other EGFR family members leading to proliferation, differentiation, antiapoptosis [44] and tumor progression [45] . Herceptin was the first genome based targeted anticancer therapeutic, approved by FDA in 1998. Binding of Herceptin to HER receptor leads to receptor internalization, inhibition of cell cycle progression and antibody dependent cellular toxicity or eliciting the immune response. Herceptin induces normalization and regression of angiogenesis in HER-2 overexpressing breast cancer [46] and even blocks cleavage of HER2, which generates a membrane-bound constitutively active truncated receptor. Rituximab is an anti CD20 antibody, effective against Non Hodkins Lymphoma [47] .
EFGR is also overexpresed in many cancers especially in breast cancer. EGFR over amplification also leads to suppression of apoptosis. Proliferation, migration, stromal cell invasion and neovascularization. Important anti-EGFR monoclonal antibodies are C225 by Imclone and 2C4 by Genetech and Osidem by Mederex. C225 or cetuximab is targeted against EGFR/ Erb B1 while 2C4 is targeted against HER-2/ Erb B2 member of the EGFR family. Angiogenesis, the formation of new blood vessels to the tumor site is mainly induced and regulated by VEGF. Blocking angiogenesis is now considered to be a promising approach for anticancer therapy and the use of anti-VEGF MAb has demonstrated tumor suppression.
Certain antigens are overexpressed in cancers. P12 antigen overexpression is associated with a wide range of cancer cell lines and tissues and antibody directed towards these antigens may serve as important contributors to cancer therapeutics as exemplified by the results of priliminary trials. MAb P12 reacts with the carbohydrate sequence present on high molecular weight glycoproteins. Oncologists are now interested in newer MAbs as promising agents for the treatment of cancer.
5.5. Hsp 90 and other novel strategies
Heat shock proteins (Hsp-s) are ubiquitous proteins known for the maintenance of cellular homeostasis and are inducible under variety of stresses. Hsp-s are mainly involved in the proper folding of other proteins and hence referred to as molecular chaperons [48,49] . The accumulation of Hsp-s is seen in pathological conditions and tumors. Most kinases require molecular chaperons to maintain their activation competent conformation. Hsp-s interacts with and stabilizes various kinases [50] . Chaperon based inhibitors other than interacting with protein kinases, prevent the associated chaperon(s) from maintaining the activation competent conformation of the kinase. The result being the proteosomal degradation of the misfolded kinases, thus diminishing the level of many kinases. The Hsp-s has an unique ATP binding site, including a Bergerat fold characteristic of bacterial gyrase, topoisomerases and histidine kinases. Thus the ATP binding site serves as a robust antitumor target for kinase related chaperone machinery. Important examples are Geldanamycin, Cisplatin, Novobiocin, Radicol and other purine based inhibitors. Geldanamycin affects ErbB2, EGF, v-Src, Raf-1 etc [51,52] .
5.6. Antibody drug conjugate
Frontline therapy being scarcely full proof, hence there is a resurgent trend towards immunotherapy. The efficacy of the antibodies that targets specific molecules expressed by tumor cells can be increased by attaching toxins to them [53] . Existing immunotoxins are based on bacterial toxins like pseudomonas exotoxin, plant exotoxin like ricin or radio-nucleotides. The toxins are chemically conjugated to a specific ligand such as the variable domain of the heavy or light chain of the monoclonal antibody. Normal cells lacking the cancer specific antigens are not targeted by the targeted antibody [54] . The most promising immunotoxin is the EGF fusion protein DAB389EGF, which is a fused EGF specific sequence and diphtheria toxin and have been found to be effective in EGFR over expressing breast tumor and non small cell lung cancer. The use of therapeutic antibody for the development of antibody-drug conjugate has been tried to improve the therapeutic potential e.g. Tositumomab by GlaxoSmithcline [55] , Anti-Tac(Fv)-PE38(LMB-2) against CD25, in B, T cell lymphoma and anti-B4-bR against CD19 in B-Non Hodgkins lymphoma [56] . As molecular studies of cancer have started revealing an increased epitope repertoire due to great strides in genomics and proteomics, the search for more effective Antibody-drug conjugate has got a spur in recent times [57] .
5.7. Antisense strategies and peptide drugs
Antisense are small pieces of synthetic oligonucleotides that are designed to interact with the mRNA by Watson-Crick base pairing to block the transcription and thus translation of target proteins. Antisense oligodeoxynucleotides (ODN) targeting IGF-1R induces apoptosis in malignant melanoma and is also effective in breast cancer [58] .
Protein–protein interaction is very important in cellular processes. Hence peptide and peptidomimetics that interfere with this interaction are important. Grb2 is an essential component in the Ras signaling pathway and it's interaction with Sos is responsible for the down stream signaling process. Proline rich “Peptidimer” having two VPPVPPRRR sequence specifically recognizes Grb2 and selectively blocks Grb2-Sos interaction an important step in EGFR over expressing cancers [31] . Small peptides mAZpTyr-(α-Me)pTyr-Asn-NH2 [58] and peptidomimetics like AHNP anti erbB2 are even known to inhibit unwanted tyrosine kinase dimerization by competing with target proteins thus the peptides or peptidomimetics acts as antagonist of receptor tyrosine kinase [59,60] .
5.8. Inhibitors of Angiogenesis
Tumor cell transformation is a multistage process. An insitu tumor after a period of time abruptly sparks the formation of new blood vessels from the preexisting vasculature a process termed as angiogenesis or neovascularization. Tyrosine kinase plays an important role in this process [61] . This process though occurs normally during embryonic development, female reproductive cycle or wound healing is found as a crucial step in tumor transition from benign to malignant form, capable of spreading throughout the body [61, 62, 63] .
VEGF (Flt-1) receptor tyrosine kinase mediated mitogenesis of human endothelial cells and growth of multiple tumor types is inhibited by SU5416 [64] . PD173074 is a potent inhibitor of FGFR1 and also VEGFR2 [65] . PD98059 is an important inhibitor of MAPK cascade that lies downstream of Ras pathway and thus is effective in many tumors [66] . Antiangiogenic drugs stops new vessels from forming around a tumor and break up the existing network of abnormal capillaries that feeds the cancerous mass, thus shrinks the tumor by limiting blood supply. Treatment of cancer faces the main fear of acquired drug resistance but antiangiogenic therapy does not induce acquired drug resistance and hence can prove valuable for long-term maintenance therapy [67] .
Schematic structure of different approaches for tyrosine kinase inhibition
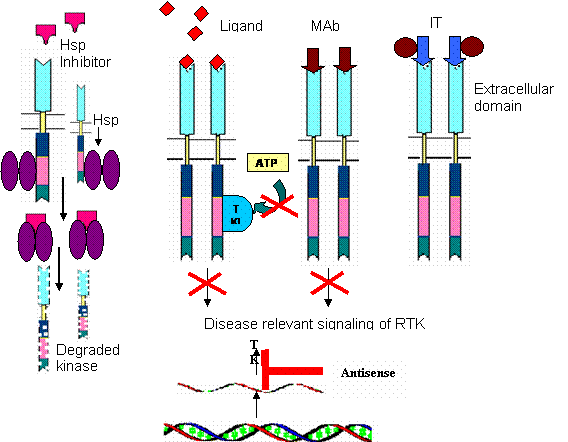
6. Mining the kinome to meet the future challenges
Cancer arises by clonal proliferation from a cell, which builds up a series of mutations leading to abnormal signaling [68] . The Achilles' heel lies in that the cancer cell depends highly on these oncogenic mutations and is said to be “oncogene addicted” [69,70] . With over 500 kinases in the human kinome and as many as 200-300 protein kinases mediating a large number of pathways in a cell at a particular time, selectivity becomes very important considering the cost of drug development. Hence strategies to move kinase drug discovery in a more rapid, comprehensive and efficient manner, including tyrosine kinase target validation, selectivity and drugability are the demand of time [71] .
Biochemical, cell based assay and screening methods for the through profiling of the kinase inhibitors using monoclonal antibody based multi-immunoblotting, fluorescent polarization assays, nonradioactive high throughput assays, 2-D NMR approaches should be exploited. Structure based drug design (SBDD) strategies depending on bioinformatics, computational approaches, mathematical models of tumor and normal tissue response, highthroughput X-ray crystallography and chemo-genomic approaches can be used to advance molecules through the routine drug discovery process. The use of recent strides in RNA interferance (RNAi) appears to be a promising approach for silencing gene expression, thus elucidating genetics of human disease with emphasis into the biological role of the kinase signaling pathways [72,73] . The promising progress made by the drugs Gleevec, Iressa and Herceptin has brought to light the potential of new innovative genome based molecular therapeutics [74]
7. Conclusion
The role of tyrosine kinase in the control of cellular growth and differentiation is central to all organisms and has been found to participate in human neoplastic diseases. Tyrosine kinase inhibitors and their potential in clinical application are well documented by dramatic examples like, Gleevec, Iressa and Herceptin. Several tyrosine kinase inhibitors are undergoing human trials and several are in the pipeline of drug discovery. The activities of these drugs are restricted to cancers with alterations in kinase targets, hence broad application of this treatment strategy are challenging. The quick selection of epidemiologically relevant, drugable tyrosine kinase targets coupled to efficient lead finding and optimization needs more intervention in the area of highthroughput cancer genome based molecular therapeutics. All these concerted effort may pave the silverlining to tailor made personalised cancer therapeutics.
Conflict of interest
The authors have declared that no conflict of interest exists.
Abbreviations
AML: acute myeloid leukemia
ALL: acute lymphocytic leukemia
CML: chronic myeloid leukemia
CMML: chronic myelomonocytic leukemia
EGF: epidermal growth factor
FGF: fibroblast growth factor
FGFR: fibroblast growth factor receptor
GIST: gastrointestinal stromal tumor
IGF: insulin like growth factor
IGFR: insulin like growth factor receptor
MAPK: mitogen activated protein kinase
PDGFR: platelet-derived growth factor receptor
PDGF: platelet-derived growth factor
PI3K: phosphatidyl inositol kinase
SH2: Src homology 2
IR: insulin receptor
mTOR: mammalian target of rapamycin
References
1. Hunter T. Signaling-2000 and Beyond. Cell. 2000 ;100 :113 -127
2. Schlessinger J. Cell Signaling by receptor tyrosine kinases. Cell. 2000 ;103 :211 -225
3. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001 ;411 :355 -365
4. Hunter T, Cooper JA. Protein-tyrosine kinases. Annu Rev Biochem. 1985 ;54 :897 -930
5. Carpenter G, King LJr, Cohen S. Epidermal growth factor stimulates phosphorylation in membrane preperations in vitro. Nature. 1978 ;276 :409 -410
6. Workman P. Paul workman on the challenges of cancer drug development. Drug Disc Today. 2003 ;8 :775 -777
7. Sawyers CL. Rational therapeutic intervention in cancer: kinases as drug targets. Curr Opin Gen Develop. 2002 ;12 :111 -115
8. Hunter T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signalling. Cell. 1995 ;80 :225 -236
9. Schenk PW, Snarr-Jagalska BE. Signal perception and transduction: the role of protein kinases. Biochim Biophys Acta. 1999 ;1449 :1 -24
10. Heldin GH. Dimerization of cell surface receptor tyrosine kinases. Cell. 1995 ;80 :213 -223
11. Bertram JS. The molecular biology of cancer. Mol Aspects Med. 2001 ;21 :167 -223
12. Nishikawa REA. A mutant epidermal growth factor receptor common in human Glioma confers enhanced tumorigenicity. Proc Natl Acad Sci. 1994 ;91 :7727 -7731
13. Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase as targets for anticancer drugs. Trends Mol Med. 2002 ;8 :17 -23
14. Buchdunger E, Matter A, Druker BJ. Bcr-Abl inhibition as a modality of CML therapeutics. Biochim Biophys Acta. 2001 ;1551 :M11 -M18
15. Kolibaba KS, Druker BJ. Protein tyrosine kinases and cancer. Biochim Biophys Acta. 1997 ;1333 :F217 -F248
16. John AM, Thomas NSB, Mufti GJ, Padua RA. Targeted therapies in myeloid leukemia. Semin Cancer Biol. 2004 ;14 :41 -62
17. Daley GQ, VanEtten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210 Bcr-Abl gene of the Philadelphia chromosome. Science. 1990 ;247 :824 -830
18. Goga A, McLaughlin J, Agar DE, Saffron DC, Witte ON. Alternative signals to RAS for the hematopoietic transformation by the BCR-ABL oncogene. Cell. 1995 ;82 :981 -988
19. Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t (5; 12) chromosomal translocation. Cell. 1994 ;77 :307 -316
20. Golub TR, Lean TM, Stegmaier K, Carroll M, Tomasson M, Gilliland DG. The TEL gene and human leukemia. Biochem Biophys Acta. 1996 ;1288 :M7 -M10
21. Shiota M, Nakamura S, Ichinohasama R. et al. Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK: a distinct clinicopathologic entity. Blood. 1995 ;86 :1954 -1960
22. Tateishi M, Ishida T, Mitsudomi T, Kaneko S, Sugimachi K. Immunohistochemical evidence of autocrine growth factors in adenocarcinoma of the human lung. Cancer Res. 1990 ;50 :7077 -7080
23. Rusch V, Baselga J, Cardon-cardo C, Orazem J, Zaman M, Hoda S Mcintosh, J Kunie, J Dmitrovsky, E . Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res. 1993 ;53 :2379 -2385
24. Wong A, Bigner S, Bigner D, Kinzler K, Hamilton S, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci. 1987 ;84 :6899 -6903
25. Smith K, Fennelly JA, Neal DE, Hall RR, Harris AL. Characterization and quantitation of the epidermal growth factor receptor in invasive and superficial bladder tumors. Cancer Res. 1989 ;49 :5810 -5815
26. Hermanson M, Funa K, Hartman H, Claesson-Welsh L, Holdin CHB Westermark, M Nister . Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992 ;52 :3213 -3219
27. Reeve JG, Morgan J, Schwander J, Bleehen NM. Role for membrane and secreted insulin-like growth factor-binding protein-2 in the regulation of insulin-like growth factor action in lung tumors. Cancer Res. 1993 ;53 :4680 -4685
28. Resnik JL, Reichart DB, Huey K, Webster NJ, Seely BL. Elevated insulin-like growth factor I receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res. 1998 ;58 :1159 -1164
29. Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, Hennekens CH, Pollak M. Plasma Insulin-Like Growth Factor-I and Prostate Cancer Risk: A Prospective Study. Science. 1998 ;279 :563 -556
30. Koppal T. Neglected kinase targets are now in vogue. Drug Disc Develop. 2003 :- Aug: 75-80
31. Bennasroune A, Gardin A, Aunis D, Cremel G, Hubert P. Tyrosine kinase receptors as attractive targets for cancer therapy. Crit Rev Oncol Hematol. 2004 ;50 (1) :23 -38
32. Hubbard SR. Protein tyrosine kinases: autoregulation and small-molecule inhibition. Curr Opin Struc Biol. 2002 ;12 :735 -741
33. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000 ;35 :95 -105
34. Fabbro D, Ruetz S, Buchdunger E, Cowan-Jacob SW, Fendrich G, Liebetanz J, Mestan J, O'Reilley T, Traxler P, Chaudhuri B, Fretz H, Zimmermann J, Meyer T, Caravatti G, Furet P, Manley PW. Protein kinases as targets for anticancer agents: from inhibitors to useful drugs. Pharmacol Ther. 2002 ;93 :79 -98
35. Yaish P, Gazit A, Gilon C, Levitzki A. Blocking of EGF-dependent cell proliferation by EGF receptor kinase inhibitors. Science. 1988 ;242 :933 -935
36. Fabbro D, Parkinson D, Matter A. Protein tyrosine kinase inhibitors: new treatment modalities? Curr Opin Pharmacol. 2002 ;2 :374 -381
37. Gazit A, Yaish P, Gilon C, Levitzki A. Tyrphostins I: synthesis and biological activity of protein tyrosine kinase inhibitors. J Med Chem. 1989 ;32 :2344 -2352
38. Levitzki A. Tyrosine kinases as targets for cancer therapy. Eur J Cancer. 2002 ;38 :S11 -S18
39. Druker BJ. STI571 (GleevecTM ) as a paradigm for cancer therapy. Trends Mol Med. 2002 ;8 :S14 -S18
40. Fukuoka M, Yano S, Giaccone G. et al. Multi-Institutional Randomized Phase II Trial of Gefitinib for Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2003 ;21 :2237 -2246
41. Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA. 2001 ;98 :10314 -10319
42. Houshmand P, Zlotnik A. Targeting tumor cells. Curr Opin Cell Biol. 2003 ;15 :640 -644
43. Craven RJ, Lightfoot H, Cance WG. A decade of tyrosine kinases: from gene discovery to therapeutics. Surgical Oncology. 2003 ;12 :39 -50
44. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/ neu oncogene. Science. 1967 ;235 :177 -182
45. Guy CT, Cardiff RD, Muller WJ. Activated neu Induces Rapid Tumor Progression. J Biol Chem. 1996 ;271 :7673 -7678
46. Izumi Y, Xu L, Tomaso ED, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002 ;416 :279 -280
47. Fischer OM, S.Treit S, Hart S, Ulrich A. Beyond Herceptin and Gleevec. Curr Opin Chem Biol. 2003 ;7 :490 -495
48. Bakau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998 ;92 :351 -356
49. Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996 ;381 (6583) :571 -579
50. Yarden Y, Sliwkowsky MX. Untangling the ErbB signaling network. Nature Rev Mol Cell Biol. 2001 ;2 :127 -137
51. Sreedhar AS, Soti C, Csermely P. Inhibition of Hsp 90: a new strategy for inhibiting protein kinases. Biochim Biophys Acta. 2003 :- (in print)
52. Workman P, Kaye SB. Translating basic cancer research into new cancer therapeutics. Trends Mol Med. 2002 ;8 :S1 -S9
53. Colaco CALS. Cancer immunotherapy: simple cell biology? Trends Mol Med. 2003 ;12 :515 -516
54. Kreitman RJ. Immunotoxins in cancer therapy. Curr Opin Immun. 1999 ;11 :570 -578
55. Cheson B. Bexxar (Corixa/Glaxo SmithKline). Curr Opin Investig Drugs. 2002 ;3 :165 -170
56. Grossbard ML, Freedman AS, Ritz J, Coral F, Goldmacher VS, Eliseo L, Spector N, Dear K, Lambert JM, Blattler WA. Serotherapy of B-cell neoplasms with anti-B4-blocked ricin: a phase I trial of daily bolus infusion. Blood. 1992 ;79 :576 -585
57. Payne G. Progress in immunoconnjugate cancer therapeutics. Cancer Cell. 2003 ;3 :207 -212
58. Andrews DW, Resnicoff M, Flanders AE, Kenyon L, Curtis M, Merli G, Baserga R, Iliakis G, Aiken RD. Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin-like growth factor type I receptor in malignant astrocytomas. J Clin Oncol. 2001 ;19 :2189 -2200
59. Liu W, Vidal M, Gresh N, Roques BP, Garbay C. Small Peptides Containing Phosphotyrosine and Adjacent αMe-Phosphotyrosine or Its Mimetics as Highly Potent Inhibitors of Grb2 SH2 Domain. J Med Chem. 1999 ;42 :3737 -3741
60. Berezov A, Chen J, Liu Q, Zhang H, Greene MI, Murali R. Disabling Receptor Ensembles with Rationally Designed Inteface Peptidomimetics. J Biol Chem. 2002 ;277 :28330 -28339
61. Cohen P. The development and therapeutic potential of protein kinase inhibitors. Curr Opin Chem Biol. 1999 ;3 :459 -465
62. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996 ;86 :353 -364
63. Kerbel RS. A cancer therapy resistant to resistance. Nature. 1997 ;390 :335 -336
64. Annie T, Fong T, Shawver LK, Sun L, Tang C, App H, Powell J, Kim YH, Schreck R, Wang X, Risan W, Ullrich Axel, Peter Hirth K, McMahon G. SU5416 Is a Potent and Selective Inhibitor of the Vascular Endothelial Growth Factor Receptor (Flk-1/KDR) That Inhibits Tyrosine Kinase Catalysis, Tumor Vascularization, and Growth of Multiple Tumor Types. Cancer Res. 1999 ;59 :99 -106
65. Mohammadi M, Frorum S, Hamby JM, Schroever MC, Panek RL, Lu GH, Eliseenkova AV, Green D, Schlessinger J, Hubbard SR. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 1998 ;17 :5896 -5904
66. Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A Synthetic Inhibitor of the Mitogen-Activated Protein Kinase Cascade. PNAS USA. 1995 ;92 :7686 -7689
67. Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997 ;27 :404 -407
68. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001 ;411 :366 -374
69. Klausner RD. The fabric of cancer cell biology-Weaving together the strands. Cancer Cell. 2002 ;1 :3 -11
70. Weinberg RA. How cancer arises? Scientific American. 1996 ;275 :32 -40
71. Gill A. Protein kinases in drug discovery and development. Drug Disc. Today. 2004 ;9 :16 -17
72. Dunn DA. Mining the human 'kinome'. Drug Disc Today. 2002 ;7 :1121 -1123
73. Hellman S, Everett E. Advancing Current Treatments for Cancer. Scientific American. 1996 ;275 :84 -89
74. Workman P. Scoring a bull's-eye against cancer genome targets. Curr Opin Pharmacol. 2001 ;1 :342 -352
75. Stahel R, Rossi A, Petruzelka L, Kosimidis P, Braud F, Bernardo MM, Souquet P-J, Soto Parra H, Gridelli C. Lessons from the 'Iressa' Expanded Access Programme: gefitinib in special non-small-cell lung cancer patient populations. British J Cancer. 2003 ;89 :S19 -S23
76. Kedar D, Baker CH, Killion JJ, Dinney CPN, Fidler IJ. Blockade of the Epidermal Growth Factor Receptor Signaling Inhibits Angiogenesis Leading to Regression of Human Renal Cell Carcinoma Growing Orthotopically in Nude Mice. Clin Cancer Res. 2002 ;8 :3592 -3600
77. Drevs J, Hofmann I, Hugenschmidt H, Wittig C, Madjar H, Muller M, Wood J, Martiny-Baron G, Unger C, Marme D. Effects of PTK787/ZK 222584, a specific inhibitor of vascular endothelial growth factor receptor tyrosine kinases, on primary tumor, metastasis, vessel density, and blood flow in a murine renal cell carcinoma model. Cancer Res. 2000 ;60 :4819 -4824
78. Lionel DL, Andrew PB, Bernard FC, Paul KW, Jan LF, Mary GW, Peter AK, Marc SE. The pharmacokinetics of the bispecific antibody MDX-H210 when combined with interferon gamma-1b in a multiple dose phase I study in patients with advanced cancer. Cancer Chemo Pharmacol. 2002 ;49 :375 -384
79. Yang XH, Cooper S, Dean N, Holmlund J, Dorr A, Monia BP. Anti-tumor activity of LY900003 (ISIS3521) in human tumor xenograft models. Proc Am Soc Clin Oncol. 2003 ;22 :215 -
80. Lockhart AC, Rudin C, Berlin J, Roth BJ, Hande KR, Martin RR, Sullivan TM, Grindel JM, Zhang R, Rothenberg RL. Gem 231, a second -generation anti-PKA oligonucleotide, administered with weekly CPT-11: A phase I trial. Proc Am Soc Clin Oncol. 2003 ;22 :245 -
81. Krietman RJ, Chaudhary VK, Waldmann T, Willingham MC, FitzGerald DJ, Pastan I. The recombinant Immunotoxin Anti-Tac(Fv)-Pseudomonas Exotoxin 40 is cytotoxic towards Peripheral Blood Malignant Cells from patients with adult T-cell leukemia. Proc Natl Acad Sci USA. 1990 ;87 :8291 -8295
82. Frankel AE, Fleming DR, Powell BL, Gartenhaus R. DAB(389)IL2 (ONTAK®) fusion protein therapy of chronic lymphocytic leukaemia. Expert Opin Biol Ther. 2003 ;3 :179 -186
83. Park JW, Yeh MW, Wong MG, Lobo M, Hyun WC, Duh QH, Clark OH. The heat shock protein 90-binding geldanamycin inhibits cell proliferation, down- regulates oncoproteins, and inhibits epidermal growth factor-induced invasion in thyroid cancer cell line. J Clin Endocrionol Metab. 2003 ;88 :3346 -3353
84. Munster PN, Srethapakdi M, Moasser MM, Rosen N. Inhibition of heat shock protein 90 function by Ansamycins causes the morphological and functional differentiation of breast cancer cells. Cancer Res. 2001 ;61 :2945 -2952
Tables and Figures
Mechanism of oncogenic activation of tyrosine kinase
| Gene | Mutation | Disease |
| EGFRvIII | Extracellular domain | Glioblastoma, Ovarian tumor, Non small cell lung carcinoma |
| FGFR3 | Extracellular domain | Multiple myeloma |
| BCR-ABL | Translocation t(9:22) | Chronic myeloid leukemia Acute lymphocytic leukemia |
| TEl-ABL | Translocation t(9:12) | Acute lymphocytic leukemia |
| TEL-PDGF | Translocation t(5:12) | Chronic myeloid monocytic leukemia |
| EGFR and EGF, TGFα | Autocrine-paracrine loop | Non small cell lung carcinoma Bladder cancer Glioblastoma multiforme |
| PDGFR and PDGF | Autocrine-paracrine loop | Glioma |
| IGFR and IGF I and II | Autocrine-paracrine loop | Breast cancer |
Protein kinase inhibitors in clinical trials
| Inhibitor | Target kinase(s) | Company | Cancer | Status | Ref. |
| Small molecule inhibitors | |||||
| STI-571 | Abl, c-kit, PDGFR | Novartis | CML, GIST | launched | [39] |
| ZD1839 | EGFR | AstraZeneca | NSCLC, SCC | launched | [40] |
| OSI-774 | EGFR | OSI/Genetech | SCC, BC, LC | Phase III | [75] |
| PKI-166 | EGFR, Her-2 | Novartis | general cancer | Phase II | [76] |
| PTK-787 | VEGFR | Novartis | CR cancer | Phase III | [77] |
| SU5416 | VEGFR | Sugen | solid tumor | Phase III | |
| Monoclonal antibody | |||||
| Herceptin | Her-2/neu | Genetech | BC | FDA approved | [46] |
| MDX-H210 | Her-2/neu | Madarex | BC | Phase I | [78] |
| 2C4 | Her-2/neu | Genetech | BC, PC, OC | Phase II | - |
| C225 | EGFR | Imclone | Pan.C, BC, RenC | Phase III | - |
| Antisense | |||||
| ISIS 3521 | PKC-α | ISIS | NSCLC, BC, Pan.C | Phase II | [79] |
| GEM 231 | PKA | Hybridon | CR, Pan.C, LC | Phase I | [80] |
| Immunotoxins | |||||
| Anti-Tac (Fv)-PE38 | CD25 | - | B, T cell leukemia | Phase I | [81] |
| DAB389IL2 | IL-2R | - | CTCL, HD, B-NHL | Phase III | [82] |
| Hsp90 inhibitors | |||||
| Geldanamycin | Hsp90 | Conforma. Inc. | Thyroid cancer | Phase I | [83] |
| 17-AAG | Hsp90 | - | BC Phase I | Phase I | [84] |
BC- Breast cancer, LC- Lung cancer, NSCLC- Non small cell lung cancer, SSC- squamous cell carcinoma, CR- Colorectal cancer, CML- Chronic myeloid leukemia, GIST- Gastrointestinial stromal tumor, CTCL-Cutaneous T cell lymphoma, NHL- Non Hodgkins lymphoma, HD- Hodgkins disease, Pan.C – Pancreatic cancer, Ren.C- Renal cancer, KHK- Kyowa Hakko Kogyo Ltd.
Author biography
Anup K. Mukhopadhyay is an Associate Professor of the Department of Biotechnology, National Institute of Pharmaceutical Education and Research, Punjab, India-160062. His research interest is in the area of mitochondrial bioenergetics of cancer cell of haemato-oncologic diseases. His research interest also includes kinetics of electron transfer enzyme and reactive oxygen species.
Manash K. Paul is a PhD student of the Department of Biotechnology, National Institute of Pharmaceutical Education and Research, Punjab, India-160062. his current research includes target enzymes in cancer therapeutics and mitochondrial bioenergetics in cancer leukocyte.
![]() Corresponding address:
Corresponding address:
Dr. Anup K. Mukhopadhyay, NIPER Communication No 283. e-mail: anupniperco.in or akmukhopadhayayac.in Fax: 91172214692 Tel: 911722214682