Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2004; 1(1):34-42. doi:10.7150/ijms.1.34 This issue Cite
Case report
The Syndrome of Frontonasal Dysplasia, Callosal Agenesis, Basal Encephalocele, and Eye Anomalies – Phenotypic and Aetiological Considerations
Antonio Richieri-Costa, Maria Leine Guion-Almeida ![]()
Hospital de Reabilitação de Anomalias Craniofaciais, Universidade de São Paulo, Bauru-SP, Brasil
Received 2004-2-16; Accepted 2004-3-3; Published 2004-3-10
Abstract
We report ten sporadic cases of Brazilian patients with facial midline defects, callosal agenesis, basal encephalocele, and ocular anomalies. This very rare cluster of anomalies has been well reported before. However, only until recently it is recognized as a syndrome belonging to frontonasal dysplasia spectrum. The ten cases confirm a distinct clinical entity and help to define the phenotype more precisely than previously. Up to now etiology remains unknown, although we conjecture that it is due to a mutation in TGIF gene.
Keywords: Morning glory syndrome, frontonasal dysplasia, CNS midline anomalies
1. Introduction
Frontonasal dysplasia (FND) is the hallmark of several syndromes involving the frontonasal process that includes: isolated frontonasal dysplasia (MIM 136760 and 305645), acrofrontofacionasal dysostosis 1 (MIM 201180), acrofrontofacionasal dysostosis 2 (MIM 201181), frontofacionasal dysplasia (MIM 229400), oculoauriculofrontonasal syndrome (MIM 601452), among other related conditions [1]. One of the syndromes within this spectrum comprises of the syndrome of midline facial defects, callosal agenesis, basal encephaloceles, and eye anomalies. It has been described under different eponyms, most commonly the morning glory syndrome. The anomalies observed in the present condition are embryological related, occurring in the same critical period of time, most likely during the early induction of the forebrain, resulting in anomalies involving frontonasal process, midfacial structures as well as the primordium of the eyes. Several genes are known to exert effects during this period resulting in different craniofacial anomalies [2]. Up to now there is no evidence of any particular gene involved in etiology of any reported condition within the FND spectrum. One of the patients here described was previously evaluated within a sample of patients purportedly presenting holoprosencephaly (HPE) [3] and showed a 485C→T mutation in exon 4 of the TGIF gene. We would like to suggest that the syndrome here reported could be related to mutation in this gene.
2. Clinical Report
We evaluated ten patients with FND, callosal agenesis, basal encephalocele, and eye anomalies syndrome ascertained through the Serviço de Genética Clínica do Hospital de Reabilitação de Anomalias Craniofaciais da Universidade de São Paulo-Bauru-SP, Brasil. All index cases were isolated with no recurrence in relatives. There were no antecedents of neural tube defect in the families. There was no parental consanguinity. The sex ratio of the sample was 1:1. Age ranged from 5 months to 15 years. Parental age was within the population average. Family history was obtained by personal interview. Pregnancy history and delivery were unremarkable. There was no reference to toxic, infectious, or teratogenic agents during the pregnancy. Clinical, laboratorial, and radiological data are presented in Table 1. Photographic documentation is shown in the back of paper.
3. Discussion
Midline craniofacial and central nervous system anomalies are commonly associated with eye anomalies [4; 5; 6; 7; 8; 9]. Callosal agenesis, basal encephaloceles, FND, and ocular anomalies may be manifested as isolated anomalies or in different combination as part of the clinical picture of some dozen of syndromes, however, this unique association represents a well-defined syndrome within the FND spectrum. In relation to this one, up to now, there are no objective correlations toward a definitive etiological agent nor definitive delineation of the phenotype since different conditions have been named within this spectrum [10; 11; 12; 13; 14]. Taking into account the above considerations, as well as the present casuistic and the number of reviewed papers, the clinical delineation of the present condition can be established as follow: callosal agenesis, basal encephloceles, mainly sphenoethmoidal type, always present. Facial and ocular anomalies are variable. The former ranges from mild hypertelorism with a median upper lip notch to median cleft involving the lip and the nose, and the later includes findings such as ptosis, orbital asymmetry, strabismus, optic disc anomalies. Morning glory anomaly, despite often referred, is not an obligatory finding. It represents an isolated ocular sign rather than a syndrome, resulting from a congenital optic disc dysplasia occurring isolated or within the clinical spectrum of some sporadic mendelian, and chromosomal syndromes [14; 15; 16; 17; 18; 19]. It is remarkable that in spite of central nervous system damage, mental status is usually normal, and only occasionally learning disabilities, behavioral and/or language problems can present [20]. Image studies reveal in all cases basal encephalocele, agenesis of the corpus callosum, and well-preserved cerebral structures and anterior commissures. The extension of the basal celes is variable, and in some instances respiratory distress can be present [21]. It is evident that the manifested phenotype results from one hit event occurring before the 8 weeks gestation since: the nerve fibers of retinal ganglion cells begin to project into the CNS at 8-10 wk [2; 22]; early callosal axon guidance towards its specific target starts at 9-12 wk [23; 24]; encephaloceles arises as earlier as at 4th to 5th post conceptual weeks [25] coincidental with the development of the frontonasal process [26].
Several genes exert effects early in embryogenesis resulting in malformation of a specific structure or different related ones depending on the developmental context. Considering midline craniofacial development, in known syndromes, most important involved genes are the SHH, TGIF, GLI2, TBX22, ZIC2, SIX3, TDGF1, among others [27; 28; 29; 30; 31; 32]. TGIF mutations in humans affect brain development resulting in different pattern of cerebral and facial manifestations ranging from normality to typical HPE phenotype. Penetrance is relatively low. Patient 1 of the present study was previously reported as having HPE [3], however, reviewing our files we collected the present sample and we conclude that he presents this particular syndrome instead HPE. It is possible that the phenotypic difference existing between these conditions could be attributed to different mutations within the gene and that the 485C-T mutation in exon 4 of the TGIF gene, detected in Patient 1, could be responsible for the particular phenotype here reported. Presently we are conducting molecular studies in order to test this hypothesis.
In conclusion, the condition reported here is clinically homogeneous, and phenotypically it can be defined as frontonasal dysplasia, callosal agenesis, basal encephalocele and ocular anomalies. Some related phenotypes should be considered in different diagnosis but excluded as main diagnosis [12] (patients 2 and 4); [11]. In view of this data we would like to suggest that the FND, callosal agenesis, basal encephalocele, and eye anomalies syndrome is due to mutation of the TGIF gene.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Guion-Almeida ML, Richieri-Costa A. Frontonasal dysplasia, macroblepharon, eyelid colobomas, ear anomalies, macrostomia, mental retardation, and CNS structural anomalies. A new syndrome? Clin Dysmorphol. 1999 ;8 :1 -4
2. Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001 ;2 :458 -68
3. Gripp KW. et al. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nature Genetics. 2000 ;25 :205 -8
4. Cohen MM Jr. et al. Hypertelorism: Interorbital growth, measurements, and pathogenetic considerations. Int J Oral Maxillofac Surg. 1995 ;24 :387 -95
5. Guion-Almeida ML, Richieri-Costa A. Alobar Holoprosencephaly Sequence, Anophthalmia, Preauricular Skin Tags, and Pulmonary Hypoplasia. A Previously Undescribed Condition. Clin Dysmorphol. 1999 ;2 (3) :19 -22
6. Guion-Almeida ML, Richieri-Costa A. New Syndrome of Growth and Mental Retardation, Structural Anomalies of the Central Nervous System and First Branchial Arch, Anophthalmia, Heminasal A/hypoplasia, and Atypical Clefting: Report on 4 Brazilian Patients. Am J Med Genet. 1999 ;87 :237 -44
7. Guion-Almeida ML, Richieri-Costa A. Frontonasal dysplasia, macroblepharon, eyelid colobomas, ear anomalies, macrostomia, mental retardation, and CNS structural anomalies: Defining the phenotype. Clin Dysmorphol. 2001 ;10 :81 -86
8. Guion-Almeida ML, Kokitsu-Nakata NM, Richieri-Costa A. Clinical variability in cerebro-oculo-nasal syndrome. Report on two additional cases. Clin Dysmorphol. 2000 ;9 :253 -57
9. Guion-Almeida ML, Machado-Paula LA, Richieri-Costa A. Heminasal aplasia: How wide is the phenotype or how many syndromes are within this spectrum? Report on six new cases. Am J Med Genet. submitted
10. Leitch RJ, Winter RM. Midline craniofacial defects and morning glory disc anomaly. A distinct clinical entity. Acta Ophthalmol Scand. 1996 ;219 :16 -19
11. Ehara H. et al. New syndrome with the Sakoda complex, bilateral anophthalmia, and cortical dysgenesis. Pediatr Neurol. 1998 ;18 :445 -51
12. Lees MM. et al. Frontonasal dysplasia with optic disc anomalies and other midline craniofacial defects: a report of six cases. Clin Dysmorphol. 1998 ;7 :157 -62
13. Hodgkins P. et al. Optic disc anomalies and frontonasal dysplasia. Br J Ophthalmol. 1998 ;82 :290 -93
14. Chen CS, David D, Hanieh A. Morning glory syndrome and basal encephalocele. Childs Nerv Syst. 2003 ;20 :87 -90
15. Traboulsi EI. et al. Aniridia, atypical iris defects, optic pit and the morning glory disc anomaly in a family. Ophthalmic Paediatr Genet. 1986 ;7 :131 -35
16. Nucci P, Mets MB, Gabianelli EB. Trisomy 4q with morning glory disc anomaly. Ophthalmic Paediatr Genet. 1990 ;11 :143 -45
17. Brodsky MC. et al. Morning glory disc anomaly in neurofibromatosis type 2. Arch Ophthalmol. 1999 ;117 :839 -41
18. Krishnan C, Roy A, Traboulsi E. Morning glory disk anomaly, choroidal coloboma, and congenital constrictive malformations of the internal carotid arteries (moyamoya disease). Ophthalmic Genet. 2000 ;21 :21 -4
19. Schneider C. et al. Clinically isolated Morning glory syndrome. J Fr Ophtalmol. 2002 ;25 :178 -81
20. Nawratzki I. et al. Bilateral morning glory syndrome with midline brain lesion in an autistic child. Metab Pediatr Syst Ophthalmol. 1985 ;8 :35 -6
21. Itakura T. et al. Bilateral morning glory syndrome associated with sphenoid encephalocele. J Neurosurg. 1992 ;77 :949 -51
22. Knecht AK, Bronner-Fraser M. Induction of the neural crest: a multigene process. Nat Rev Genet. 2002 ;3 :453 -61
23. Utsunomiya H. et al. Dysgenesis of the corpus callosum and associated telencephalic anomalies: MRI. Neuroradiology. 1997 ;39 :302 -10
24. Gelot A, Esperandieu O, Pompidou A. Histogenesis of the corpus callosum. Neurochirurgie. 1998 ;44 (1 Suppl) :61 -73
25. Hoving EW. Nasal encephaloceles. Childs Nerv Syst. 2000 ;16 :702 -6
26. McBratney BM. et al. Frontonasal dysplasia in 3H1 Br/Br mice. Anat Rec. 2003 ;271A :291 -302
27. Wallis DE, Muenke M. Molecular mechanisms of holoprosencephaly. Mol Genet Metab. 1999 ;68 :126 -38
28. Chen CP. et al. Molecular diagnosis of a novel heterozygous 268C→T (R90C) mutation in TGIF gene in a fetus with holoprosencephaly and premaxillary agenesis. Prenat Diagn. 2002 ;22 :5 -7
29. de la Cruz JM. et al. A loss-of-function mutation in the CFC domain of TDGF1 is associated with human forebrain defects. Hum Genet. 2002 ;110 :422 -28
30. Aguilella C. et al. Molecular screening of the TGIF gene in holoprosencephaly: identification of two novel mutations. Hum Genet. 2003 ;112 :131 -34
31. Goodman FR. Congenital abnormalities of body patterning: embryology revisited. Lancet. 2003 ;362 :651 -62
32. Roessler E. et al. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci U S A. 2003 ;100 :13424 -429
Tables and Figures
Clinical findings of studied cases
| Patient | Sex | Age | Cleft | Ocular Anomalies | CNS Anomalies | Additional data | Laboratorial |
| 1 | M | 6 y | Midline CLP | Right palpebral ptosis and cataract | Sphenoethmoidal encephaloc ele ACC | Normal development Short stature | 46,XY TGIF gene mutation |
| 2 | M | 11 y | Midline CL Cleft uvula | Left palpebral ptosis, microcornea, and optic nerve atrophy | Sphenoethmoidal encephalocele ACC | Normal development Short stature Right choanal atresia | 46,XY |
| 3 | F | 9 y | Midline CL Bifid nasal tip | Right palpebral ptosis, microphthalmia, convergernt strabismus, and cataract | Sphenoethmoidal encephalocele ACC | Normal development | 46,XX |
| 4 | F | 15 y | Midline CLP | Bilateral palpebral ptosis | Sphenoethmoidal encephalocele ACC | Normal development | 46,XX |
| 5 | M | 13 y | Midline CLP | Left palpebral ptosis,optic bulb atrophy, and cataract Right papillary coloboma | Sphenoethmoidal encephalocele ACC | Normal development Short stature | 46,XY |
| 6 | F | 7 y | Midline CL | Left palpebral ptosis | Sphenoethmoidal encephalocele ACC | Normal development Short stature | 46,XX,r(21) |
| 7 | M | 1 y 2 m | Midline CL | miopia | Sphenoethmoidal encephalocele ACC | Mild developmental delay Growth retardation Nocturnal apnea | 46,XY |
| 8 | F | 2 y 2m | Midline CL Bifid nasal tip | Ptosis | Sphenoethmoidal encephalocele ACC | Normal development | 46,XX,9ph |
| 9 | F | 7 m | Midline CL | Right globe<left globe Left peripupilar iris atrophy, posterior polar cataract, optic nerve coloboma | Ethmoidal encephlalocele ACC | Normal development Growth retardation | 46,XX |
| 10 | M | 5 m | Midline CLP CP | Ptosis | Callosal agenesis | Nomal development Growth retardation | 46,XY |
Legend: F: female; M: male; y: years; m: months; CL: cleft lip; CP: cleft palate; CLP: cleft lip and palate; ACC: corpus callosum agenesis
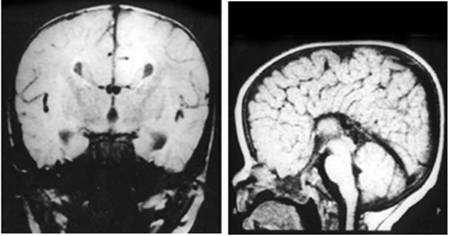
Clinical and image aspects of patient 1
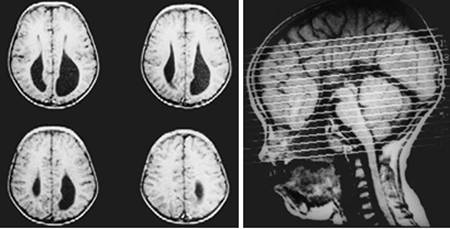
Clinical and image aspects of patient 2
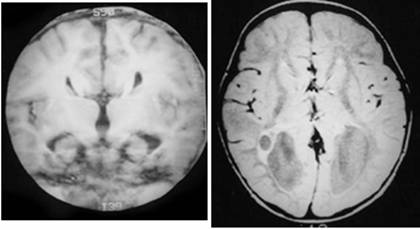
Clinical and image aspects of patient 3
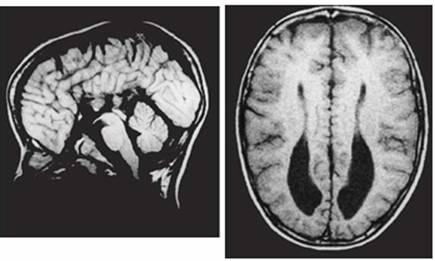
Clinical and image aspects of patient 4
Clinical and image aspects of patient 5
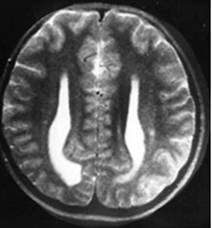
Clinical and image aspects of patient 6
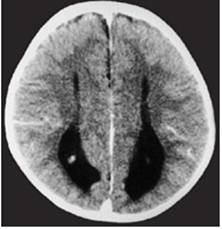
Clinical and image aspects of patient 7
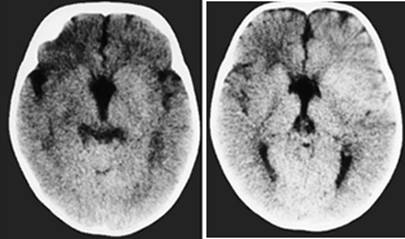
Clinical and image aspects of patient 8
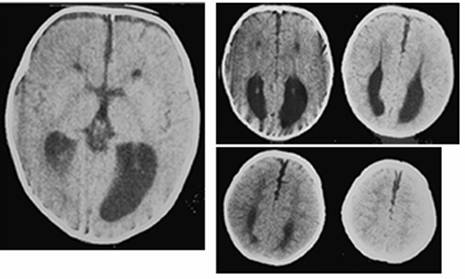
Clinical and image aspects of patient 9
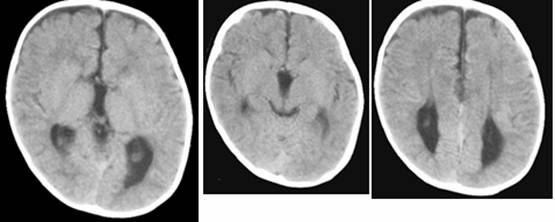
Clinical and image aspects of patient 10
Author biography
Antonio Richieri-Costa is a clinical geneticist at the HRAC-USP. His current research includes genetics in craniofacial malformations.
Maria Leine Guion-Almeida is a clinical geneticist at the HRAC-USP. Her current research includes genetics in craniofacial malformations.
![]() Corresponding address:
Corresponding address:
Antonio Richieri-Costa, MD, Hospital de Reabilitação de Anomalias Craniofaciais, Universidade de São Paulo, Rua Silvio Marchione 3-20, CEP 17012-230, SP, Bauru, Brasil. Phone: (Int+55) (14) 3235 8183. Fax: (Int+55) (14) 3234 7818. Email: richieribr