Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2026; 23(8):2621-2636. doi:10.7150/ijms.129345 This issue Cite
Research Paper
Multi-Omics and Clinical Data Analyses of Protein Arginine Methyltransferases in Pan-Cancer and Colorectal Cancer
Jialing Xie1#, Qihui Wu2,3#, Yuanyuan Xu1#, Jiaxin Liu4, Yan Wang1, Xuan Wang1, Xiaodan Fu3,5 ![]() , Yimin Li1
, Yimin Li1 ![]()
1. Department of Pathology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200025, China.
2. Department of Gynecology, Xiangya Hospital, Central South University, Changsha 410008, China.
3. National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Changsha 410008, China.
4. Department of Pathology, School of Basic Medical Sciences, Central South University, Changsha 410078, China.
5. Department of Pathology, Xiangya Hospital, Central South University, Changsha 410008, China.
# These authors have contributed equally to this work.
Received 2025-12-2; Accepted 2026-6-1; Published 2026-7-13
Abstract

Objective: Arginine methylation, catalyzed by protein arginine methyltransferases (PRMTs), is a critical post-translational modification that modulates gene expression and signal transduction. Although the involvement of PRMTs in malignancy is increasingly recognized, a comprehensive pan-cancer synthesis of this family remains elusive. We sought to elucidate the multi-omic landscape of PRMTs and define their biological contributions to colorectal cancer (CRC) progression.
Methods: Using multi-omic data from TCGA and GTEx, we performed an integrative pan-cancer analysis of PRMT expression, genetic alterations, epigenetic modifications, and their associations with prognosis, genomic heterogeneity, stemness, the tumor microenvironment, and oncogenic pathways. After validating PRMT expression patterns across multiple independent CRC cohorts from the GEO database, PRMT1 was prioritized for further investigation. Its expression was assessed in a real-world CRC cohort using immunohistochemistry, while its oncogenic potential was scrutinized through in vitro proliferation assays.
Results: PRMT family genes exhibit heterogeneous molecular profiles across cancers, which correlate significantly with genomic instability, stemness, immune infiltration, and oncogenic pathway activation. Clinical validation demonstrated that PRMT1 is markedly upregulated in CRC, where its overexpression correlates with larger tumor size, advanced T stage, and lymph node metastasis. Loss-of-function experiments demonstrated that PRMT1 is essential for maintaining CRC cell proliferation.
Conclusion: Our study highlights the multifaceted roles of PRMTs across the pan-cancer spectrum. By identifying PRMT1 as a key driver of CRC progression, this work provides a rationale for the development of PRMT-targeted therapies and personalized diagnostic biomarkers.
Keywords: PRMTs, pan-cancer, colorectal cancer, genomic heterogeneity, tumor microenvironment, prognostic
Introduction
Cancer progression is driven by complex genetic and epigenetic alterations, posing persistent challenges for diagnosis and treatment [1, 2]. Deciphering the epigenetic landscape across diverse malignancies is essential to identifying novel regulatory hubs [3, 4]. Within this realm, protein arginine methyltransferases (PRMTs) have emerged as pivotal orchestrators of tumor biology [5]. By utilizing S-adenosyl-L-methionine as a methyl donor, PRMTs catalyze the arginine methylation of diverse substrates—including histones (H3/H4) and non-histone proteins (e.g., Stat1, hnRNP)—thereby modulating fundamental cellular processes such as transcription, RNA splicing, and signal transduction [5-8]. The PRMT family, comprising multiple members with distinct substrate specificities, plays an indispensable role in both physiological homeostasis and pathological progression [9-11].
Given their central role in epigenetic regulation, the dysregulation of PRMTs has been implicated in a broad spectrum of cancers; however, the precise oncogenic mechanisms underlying their action remain to be fully elucidated [12]. While the overexpression or aberrant splicing of individual members, such as PRMT1, has been documented in breast, lung, and colorectal cancers (CRC) [13-16], most studies focus on isolated PRMT members or specific tumor types. This fragmented approach limits our understanding of their universal versus context-dependent functions. Although pan-cancer analyses offer a powerful tool to identify shared molecular signatures [17], existing family-wide investigations have largely been restricted to PRMT1, 4, and 5 [18, 19], underscoring an urgent need for a systematic, family-wide characterization across the human cancer spectrum.
Among these malignancies, CRC remains a global health burden, ranking as the third most prevalent cancer and the second leading cause of cancer-related mortality [20, 21]. Despite advances in screening, there is a critical need for novel biomarkers and therapeutic targets [22-25]. Aberrant expression of PRMTs has been shown to drive CRC growth and metastasis through multiple mechanisms [26-30]. Furthermore, the potent anti-tumor efficacy of emerging PRMT inhibitors in CRC models highlights their potential as both therapeutic agents and biomarkers for disease monitoring [31-33]. Therefore, investigating PRMTs in CRC offers both mechanistic insights into tumorigenesis and a robust platform to validate pan-cancer findings within a clinically relevant framework.
In the present study, we integrated multi-omic data from TCGA and GTEx to provide a family-wide pan-cancer profile of PRMTs. We then focused on CRC, validating our findings using GEO datasets and a real-world clinical cohort. Through immunohistochemistry and in vitro assays, we specifically investigated the clinical relevance and functional role of PRMT1. Our findings provide a comprehensive landscape of PRMTs and highlight their potential as biomarkers and therapeutic targets in precision oncology.
Materials and Methods
Data Retrieval
To characterize the molecular and functional landscape of the PRMT family, we integrated pan-cancer transcriptomic, clinical, and multi-omic data (somatic mutations, CNVs, and DNA methylation) from TCGA and GTEx via the UCSC Xena platform. The transcriptomic signatures of PRMTs were further validated in CRC using six independent GEO cohorts (GSE9394, GSE21510, GSE32323, GSE33113, GSE39582, and GSE110224). To define their roles in clinical therapy, we further curated GEO datasets of patients receiving fluoropyrimidine-based adjuvant chemotherapy (ACT, such as FOLFOX/FOLFIRI) either alone (GSE19860, GSE28702, GSE45404, GSE62080, GSE69657, and GSE72970) or in combination with bevacizumab (GSE19860, GSE19862, and GSE72970). The single-cell RNA-seq (scRNA-seq) landscape of CRC (EMTAB8107, GSE108989, GSE112865, GSE120909, GSE122969, GSE1366397, GSE139555, GSE146771, GSE166555, and GSE179784) were obtained from the TISCH2 database (http://tisch.comp-genomics.org/home/). Furthermore, the functional necessity of PRMTs for cancer cell survival was assessed by analyzing CRISPR-Cas9 dependency scores from the DepMap portal (https://depmap.org/portal/).
Human CRC Tissue Samples
Primary CRC specimens and paired adjacent normal tissues (≥ 5 cm from the tumor margin) were collected from treatment-naive patients at Xiangya Hospital. Inclusion required pathological confirmation of CRC and no history of neoadjuvant therapy or other malignancies. To ensure sample quality, tissues stored for over five years were excluded. A tissue microarray (TMA) consisting of 120 formalin-fixed, paraffin-embedded CRC tissues and 51 control tissues was employed for IHC analysis. Ethical approval and informed consent were obtained for the use of all clinical specimens.
Functional Enrichment Analysis
To dissect the signaling pathways associated with PRMT expression across various cancers, patients were stratified into high- and low-expression groups based on the top and bottom 30% of each gene's expression levels, respectively. Differentially expressed genes between these cohorts were identified using the “limma” R package, applying a threshold of |log2 fold change| ≥ 1.0. Subsequently, Gene Set Enrichment Analysis (GSEA) was conducted via the “clusterProfiler” R package, referencing the Hallmark gene sets from the Molecular Signatures Database (MSigDB, v7.5.1) [34].
Survival Analysis
The association between PRMT expression and patient prognosis was systematically evaluated using univariate Cox proportional hazards regression models across 33 TCGA cancer types. Four clinical endpoints—overall survival (OS), disease-specific survival (DSS), disease-free interval (DFI), and progression-free interval (PFI)—were included to provide a comprehensive assessment of their clinical relevance. Genes with a hazard ratio (HR) > 1 and P < 0.05 were defined as prognostic risk factors, whereas those with HR < 1 and P < 0.05 were identified as protective factors. Genes failing to meet the significance threshold (P ≥ 0.05) were deemed to have no significant impact on survival.
Tumor Microenvironment (TME) Characterization
To evaluate the TME landscape, we utilized the ESTIMATE algorithm to calculate stromal, immune, and ESTIMATE scores, as well as tumor purity, across the pan-cancer cohort [35]. The infiltration levels of various immune cell populations were quantified using the MCP-counter method [36]. Furthermore, to dissect the TME-associated functional landscape, the IOBR package [37] was leveraged to assess several biological signatures, including immune activation, stromal remodeling, cell proliferation, and DNA damage repair (DDR). We then performed correlation analyses between PRMTs expression and these functional scores. Finally, we examined the expression of six classical immune checkpoints to further elucidate the immunomodulatory role of PRMTs.
Cell Culture and RNA Interference
Human CRC cell lines (CACO2, HCT116, HCT8, HT29, LOVO, RKO, SW480, and SW620) and the normal colonic epithelial cell line NCM460 were obtained from the American Type Culture Collection. Cells were cultured in RPMI 1640 medium (Biological Industries, Israel) supplemented with 10% fetal bovine serum (Gibco, USA) and 100 U/mL penicillin-streptomycin (Invitrogen, USA) at 37°C in a humidified incubator with 5% CO2. For RNA interference, PRMT1-specific small interfering RNAs (siRNAs) were custom-synthesized by RiboBio (Guangzhou, China). The siRNA sequences were as follows: si#1, 5'-GCAACTCCATGTTTCATAA-3'; si#2, 5'-AGACGGTGTTCTACATGGA-3'. Transfections were performed using Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer's protocol.
RNA Extraction and Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from CRC cell lines using TRIzol reagent (Vazyme, Nanjing, China). cDNA was synthesized using the GoScript Reverse Transcription System (Promega, Madison, WI, USA) according to the manufacturer's protocol. qRT-PCR was performed with GoTaq qPCR Master Mix (Promega) on an ABI Prism 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Relative gene expression levels were calculated using the 2-ΔΔCT method, with GAPDH serving as the internal control. The primer sequences used were as follows: PRMT1 forward, 5'-ACTTTGACTCCTACGCACACTTTG-3' and reverse, 5'-TCCAGCACCACCTTGTCCTTG-3'; GAPDH forward, 5'-AACGGATTTGGTCGTATTGG-3' and reverse, 5'-TTGATTTTGGAGGGATCTCG-3'.
Western Blot Analysis
Total protein was extracted from cells using RIPA lysis buffer (supplemented with protease inhibitors). Equal amounts of protein were resolved by 10% SDS-PAGE and subsequently transferred onto PVDF membranes (Millipore, USA). After blocking with 5% non-fat milk for 1 h at room temperature, the membranes were probed overnight at 4 °C with primary antibodies against PRMT1 (1:1000, A1055, Abclonal, China) and GAPDH (1:2000, UM4002, Utibody, China). Following three washes with TBST, the membranes were incubated with appropriate HRP-conjugated secondary antibodies for 2 h at room temperature. Immunoreactive bands were visualized using an enhanced chemiluminescence kit.
Immunohistochemical Analysis
IHC was performed following a previously established protocol [38]. Briefly, 4-μm-thick CRC TMAs underwent dewaxing and hydration using xylene and a gradient of ethanol concentrations. Antigen retrieval involved microwave heating in sodium citrate buffer for 18 minutes. Endogenous peroxidase activity was blocked with 3% H2O2 for 10 minutes, and non-specific staining was reduced using 5% BSA. Anti-PRMT1 (1:500, A1055, Abclonal, China) was applied dropwise, followed by overnight incubation at 4 °C. A 20-minute incubation with anti-mouse/rabbit labeled polymer ensued. The exposed protein was labeled using a 0.01% DAB chromogenic solution, with nuclei counterstained using hematoxylin. Staining evaluation was independently performed by two pathologists blinded to clinicopathological information.
CCK-8 Assay
Twenty-four hours post-transfection with siRNAs (si-NC, si-#1, and si-#2), cells were trypsinized and seeded into 96-well plates at a density of 3,000 cells/well. After an additional 24-hour incubation to allow for cell attachment, cell viability was evaluated using a CCK-8 assay kit (Dojindo, Kumamoto, Japan) according to the manufacturer's instructions. Briefly, CCK-8 reagent was diluted 1:10 with fresh culture medium and added to each well. After incubation at 37 °C for 2 hours, the absorbance at 450 nm was measured using a microplate reader. To monitor cell growth over time, this procedure was repeated at 24, 48, 72, and 96 hours post-seeding.
Clonogenic Formation Assay
Following siRNA transfection, cells were harvested, resuspended, and seeded at a density of 600 cells/well in 12-well plates. The culture medium was refreshed every 2-3 days. After one week, the cell colonies were fixed with 4% paraformaldehyde for 20 minutes, and subsequently stained with 0.1% crystal violet solution for 30 minutes. After washing to remove excess dye, the plates were air-dried and photographed. The number of colonies was then quantified for further analysis.
EdU Proliferation Assay
Cell proliferation was also assessed using the BeyoClick™ EdU 594 Cell Proliferation Detection Kit (Beyotime, Shanghai, China). Briefly, 24 hours post-transfection, cells were pulse-labeled with 10 μM EdU for 2 hours. Following incubation, cells were fixed with 4% paraformaldehyde for 15 minutes and permeabilized with 0.1% Triton X-100 for 10 minutes. The click reaction was then performed in the dark for 30 minutes according to the manufacturer's instructions. Nuclei were counterstained with Hoechst 33342. Fluorescence images were captured using a fluorescence microscope to quantify the percentage of EdU-positive cells.
Statistical Analysis
All statistical analyses were conducted using the R (version 4.3.1, https://www.r-project.org/) and GraphPad Prism 9. For normally distributed variables, differences between two groups were assessed using Student's t-test, and multiple groups were compared via one-way ANOVA. Non-normally distributed variables were analyzed using the Wilcoxon test for two groups and the Kruskal-Wallis test for multiple groups. Pearson correlation analysis was utilized to calculate correlation coefficients. Heatmaps were generated using the “ComplexHeatmap” package. Results were reported as Mean ± SD. All statistical tests were two-tailed, and a P-value < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Results
Expression Profiling of PRMTs Across Pan-Cancer
To provide a foundation for our study, we first summarized the classification and catalytic mechanisms of the PRMT family (Figure 1A). Based on their specific catalytic products, PRMTs are categorized into three types: Type I (PRMT1-4, 6, and 8), which generate monomethylarginine (MMA) and asymmetric dimethylarginine (aDMA); Type II (PRMT5 and PRMT9), which produce MMA and symmetric dimethylarginine (sDMA); and Type III (PRMT7), which exclusively catalyzes the formation of MMA (Figure 1A).
Characterization and expression analysis of PRMTs across human cancers. (A) Schematic representation of protein arginine methylation, illustrating the classification and catalytic mechanisms of the three PRMT types on arginine residues. (B) Differential mRNA expression of PRMTs across TCGA and GTEx cohorts. Fold changes and P values were determined by comparing tumor tissues (TCGA) with their corresponding normal counterparts (GTEx and TCGA). Heatmap colors represent the magnitude of fold change; “Nonsense” denotes P value > 0.05.
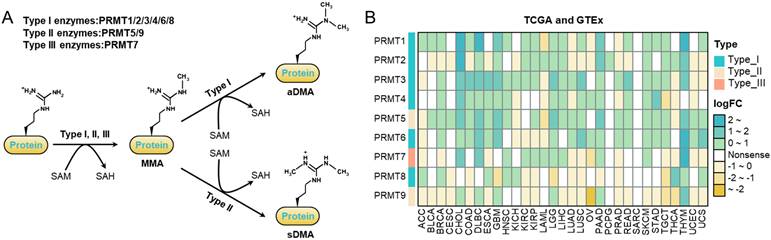
Leveraging TCGA and GTEx datasets, pan-cancer transcriptomic analysis revealed widespread dysregulation of PRMTs across various malignancies (Figure 1B, S1A-B). Most members, particularly PRMT1 and PRMT3-7, showed significant and consistent overexpression across diverse tumor types, with prominent upregulation observed in gastrointestinal and thoracic cancers (Figure 1B, S1A-B). In contrast, PRMT2 and PRMT9 displayed a distinct downregulation pattern in specific cancers, such as ovarian serous cystadenocarcinoma (OV), suggesting potential functional divergence within the PRMT family (Figure 1B, S1A-B).
Genetic Alterations and Epigenetic Regulation of PRMTs in Pan-Cancer
To characterize the genetic alterations of PRMTs, we analyzed single nucleotide variations (SNVs) across pan-cancer cohorts. The highest mutation frequency was observed in UCEC, particularly affecting PRMT3, 8, and 9, with missense mutations representing the predominant variant type (Figure 2A). In contrast, PRMTs displayed low mutation frequencies in most other tumor types (Figure 2A). Lollipop plots further revealed that these mutations were stochastically distributed along the protein sequences, lacking significant enrichment within specific functional domains (Figure S2A). Taken together, the low overall mutation frequency and sporadic distribution suggest that PRMT-driven oncogenesis is likely orchestrated by transcriptional or epigenetic dysregulation rather than recurrent driver mutations.
Genetic and epigenetic landscape of PRMTs across pan-cancer. (A) Mutational profile of PRMTs across cancer types. The left heatmap displays the number of mutated samples for each PRMT gene; '0' denotes no mutations within coding regions, and blank cells indicate no detected mutations. The right stacked bar chart summarizes the distribution of specific variant types across the PRMT family. (B-C) Correlation of PRMT mRNA expression with (B) CNVs and (C) DNA methylation. Circle size and color represent the absolute correlation coefficient and the correlation value, respectively, while the background color of each cell indicates statistical significance (P value).

Aberrant mRNA expression in cancer is frequently governed by a synergy of genomic instability and epigenetic modifications, with CNVs and DNA methylation acting as primary regulatory determinants. To further investigate the mechanisms underlying PRMT dysregulation, we first assessed the frequency of CNV alterations across various malignancies. Our results revealed that PRMT genes exhibited a relatively high frequency of CNV alterations (≥5% of samples) in most cancer types (Figure S2B). Subsequent correlation analyses showed a significant positive association between CNV levels and mRNA expression for these genes in the majority of tumors, with notable exceptions in DLBC, KICH, LAML, and THCA (Figure 2B). These findings suggest that CNVs play a major role in the aberrant expression of PRMTs and may contribute significantly to cancer progression.
Beyond structural genomic alterations, DNA promoter methylation is a key epigenetic regulator of gene transcription. Evaluation of the DNA methylation landscapes of PRMTs showed that significant divergence between tumor and normal tissues was largely absent across the family, with PRMT8 being the notable exception (Figure S2C). In line with this, correlation analysis confirmed that promoter methylation levels rarely tracked with mRNA expression in most cancers (Figure 2C). These results indicate that DNA methylation is likely not the predominant driver of PRMT dysregulation.
Prognostic Implications of PRMTs in Pan-Cancer
To further evaluate the clinical significance of PRMTs, we examined the correlation between their expression profiles and patient prognosis, focusing on OS, DSS, DFI, and PFI. Univariate Cox regression analysis of the TCGA pan-cancer dataset revealed that while the prognostic values of PRMTs were highly heterogeneous across diverse malignancies, a discernible, albeit modest, consistency in prognostic orientation was observed within specific tumor types (Figure 3). Specifically, instead of acting as isolated factors, PRMT family members often exhibited a unified trend in their impact on patient survival within the same cancer context. For instance, in adrenocortical carcinoma (ACC), elevated expression of PRMT1-5 tended to associate with unfavorable clinical outcomes, particularly shortened OS and PFI (Figure 3). Conversely, in kidney renal clear cell carcinoma (KIRC), reduced expression of PRMT2, 5, 6, 7, and 9 showed a general correlation with poorer survival, suggesting a potentially protective role for these genes in this specific malignancy (Figure 3).
Pan-cancer Prognostic Significance of PRMTs. Heatmap illustrating the associations between PRMT expression and clinical outcomes—including OS, DSS, DFI, and PFI—across 32 TCGA cancer types, as determined by univariate Cox regression analysis. Turquoise denotes a risk factor (unfavorable prognosis), whereas yellow denotes a protective factor (favorable prognosis). White indicates no statistical significance, and “N/A” represents unavailable data.

Furthermore, we investigated the prognostic impact of individual PRMTs across the pan-cancer spectrum. While PRMT1-4 frequently leaned toward being poor prognostic indicators in a wide range of cancers, PRMT5-7 exhibited dual roles, with their prognostic effects varying significantly depending on the cancer type (Figure 3). Collectively, these findings highlight the complex and context-dependent prognostic roles of PRMTs across the pan-cancer landscape, underscoring the need for further functional studies to elucidate their divergent roles in tumor biology.
Correlation of PRMTs with Genomic Heterogeneity and Stemness Indices
The prognostic impact of PRMTs prompted us to explore their links to genomic instability—a driver of tumor evolution and therapeutic resistance. Systemic analysis revealed that PRMT1, 3, 4, and 5 expression positively correlated with HRD, LOH, and ploidy across various cancers (e.g., KICH, LIHC, LUAD, LUSC), suggesting their role in exacerbating chromosomal aberrations (Figure S3). Regarding mutational landscape, the PRMT family exhibited a pro-mutational profile. Specifically, PRMT1, 4, and 5 levels were linked to increased TMB and neoantigen counts in several cancers (ACC, HNSC, LUAD, etc.) (Figure S3). Notably, PRMT1/8 (in KICH) and PRMT4/7 (in UVM) showed significant positive correlations with MSI status, potentially implying their involvement in the dysregulation of DNA mismatch repair pathways (Figure S3). Furthermore, positive correlations between PRMT1, 3, 5 and MATH scores indicated higher intratumoral heterogeneity and subclonal complexity (Figure S3). Intriguingly, PRMT expression positively correlated with tumor purity in most cases (Figure S3). This confirms that PRMTs are primarily tumor-intrinsic and ensures that the observed genomic heterogeneity is not confounded by stromal cell dilution.
As tumor stem-like cells are pivotal to heterogeneity and therapy resistance, we next assessed the association between PRMT expression and tumor dedifferentiation using six stemness indices (Figure S4). While PRMT1, 3, 4, and 5 positively correlated with stemness across multiple cancers, PRMT2 exhibited a predominantly negative correlation, highlighting functional divergence within the family (Figure S4). Interestingly, the correlations were more pronounced with RNAss (transcriptome-based) than with epigenetic indices (e.g., DNAss, DMPss) (Figure S4), suggesting that PRMTs influence stemness mainly at the transcriptional and post-transcriptional levels. These findings imply that PRMTs may drive the dedifferentiation landscape by modulating the methylation of key transcription or splicing factors.
Deciphering the Relationship between PRMTs and Tumor Microenvironment Across Pan-Cancer
Cancer progression is governed by “immunoediting,” where the immune system either eliminates malignant cells or fosters a tumor-permissive microenvironment [39]. Utilizing the ESTIMATE algorithm, we identified that most PRMTs (PRMT1, 3-9) are negatively associated with immune/stromal scores and positively with tumor purity, especially in GBM, STAD, TGCT, and THCA (Figures 4). However, PRMT2 emerged as an outlier, showing positive correlations with immune/stromal scores in COAD, DLBC, and READ (Figure 4).
Associations of PRMTs with TME scores across pan-cancer. Heatmap showing the correlation of PRMT expression with immune, stromal, and ESTIMATE scores, as well as tumor purity. Turquoise and yellow represent positive and negative correlations, respectively, while white indicates a lack of statistical significance.
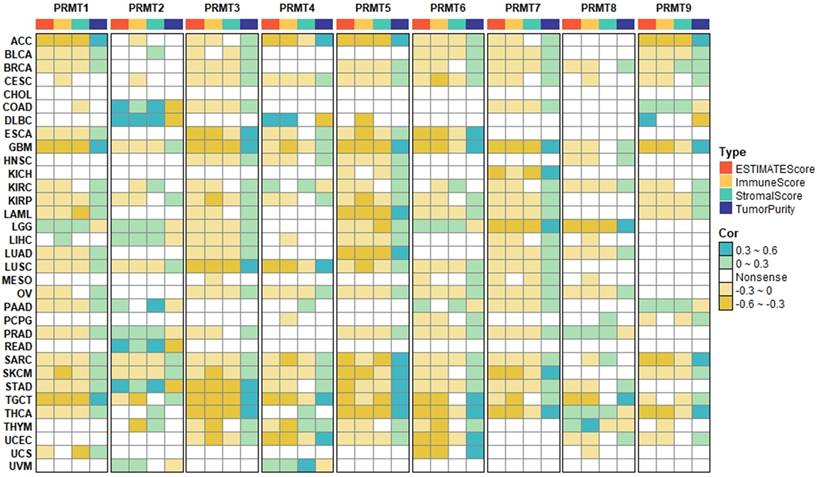
Subsequent lineage-specific quantification via MCP-counter revealed that while PRMT1, 3, and 5 inversely correlated with anti-tumor effectors (CD8⁺ T, cytotoxic lymphocytes, and NK cells), PRMT2 expression positively associated with these cells as well as stromal and myeloid components (Figure S5). Considering that PRMT2 is frequently downregulated in tumors (Figures 1B, S1A-B), its loss likely drives a transition toward an “immune-cold” state. Notably, MCP-counter provided a more nuanced view for PRMT4, 6, and 9 than the aggregate ESTIMATE scores, uncovering positive associations with endothelial cells and fibroblasts despite negative global trends (Figures 4, S5).
To further dissect the molecular underpinnings of PRMT-mediated TME remodeling, we evaluated the correlations between PRMT expression and several curated functional signatures, encompassing immune activation, stromal remodeling, and tumor-intrinsic proliferative processes. The results demonstrated that PRMT1, 3, 4, 5, 6, and 7 were robustly associated with cell-cycle progression and DDR pathways, while exhibiting modest inverse correlations with immune activation and stromal remodeling signatures (Figure S6). This underscores their primary role in maintaining genomic integrity and driving oncogenic proliferation. Conversely, PRMT2 displayed a unique functional profile, correlating positively with immune activation and stromal remodeling markers across a subset of malignancies (Figure S6). Consistently, further analysis revealed that PRMT2 was also positively associated with the expression of multiple immune checkpoints, further distinguishing it from other family members (Figure S7).
Correlation Between PRMTs and Hallmark Pathways in Pan-Cancer
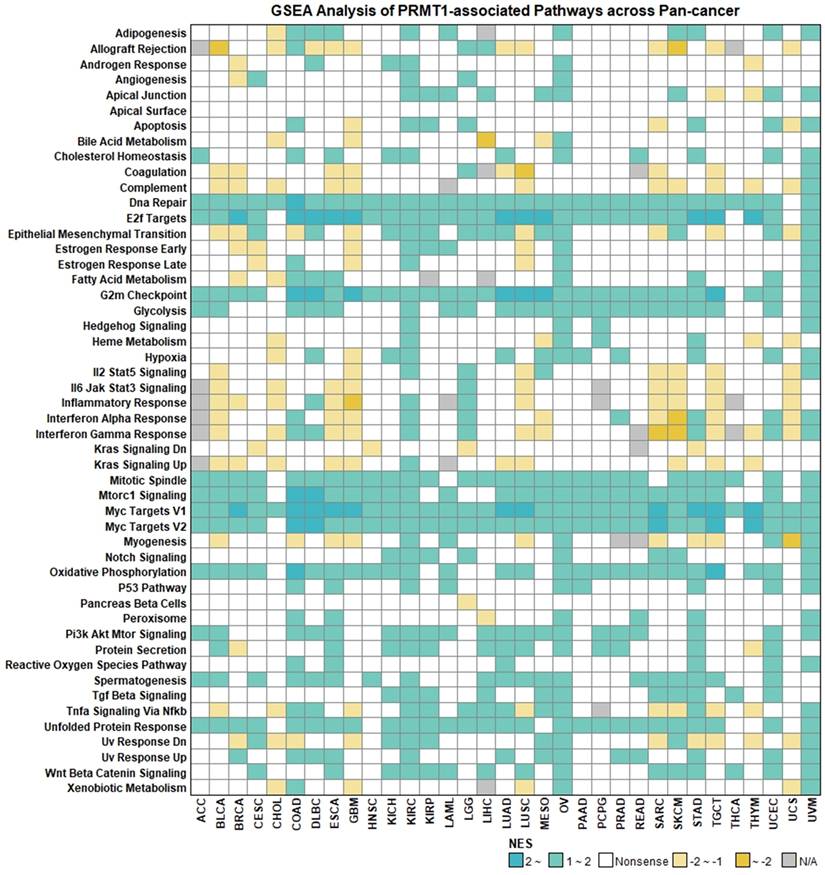
To broaden our understanding of the biological roles of PRMTs, we performed GSEA to identify enriched Hallmark pathways across pan-cancer datasets. This profiling aimed to consolidate our previous findings regarding the cell cycle and DDR while unearthing novel signaling axes through which PRMTs might modulate tumor progression and the immune landscape. Recapitulating our earlier observations, we found that RMT1, 3, 5, 6, 7, and 9 predominantly activated oncogenic and proliferative pathways, but were negatively coupled with immune-related signatures (Figure S8). Using PRMT1 as a representative model, high expression was significantly enriched in pathways governing cell-cycle progression and metabolic fitness, including DNA repair, E2F targets, G2M checkpoint, mTORC1 signaling, MYC targets, and oxidative phosphorylation (Figure 5). Furthermore, PRMT1 displayed positive associations with pro-survival cascades such as PI3K/AKT/mTOR and Wnt/β-catenin (Figure 5). In contrast, PRMT1 expression was inversely correlated with multiple immuno-stimulatory and pro-inflammatory signatures, including allograft rejection, IL2/STAT5, interferon-α/γ responses, and the IL6/JAK/STAT3 axis (Figure 5). Notably, PRMT2 exhibited a functional antithesis to its counterparts, positively correlating with immune-regulatory and inflammatory pathways (Figure S8), reinforcing its unique role in the TME.
Associations between PRMT1 and Hallmark Pathways across Pan-Cancer. Colors represent the normalized enrichment score (NES) for each pathway, and “Nonsense” indicates P > 0.05.
To determine whether the oncogenic correlations of PRMTs translate into functional requirements for survival, we assessed their genetic dependency using DepMap CRISPR screening data. While a score of 0 indicates non-essentiality, a score ≤ -1 represents core essentiality [40, 41]. We found that PRMT1 and PRMT5 are critical for cell growth across most cancer lines (Figure S9), reflecting their potent oncogenic roles. Interestingly, despite their association with proliferative programs in GSEA, PRMT3, 6, 7, and 9 yielded dependency scores near zero (Figures S8, S9). This suggests that these PRMTs may be functionally redundant or serve as context-specific modulators rather than indispensable survival factors.
The Expression Landscape of PRMTs in CRC
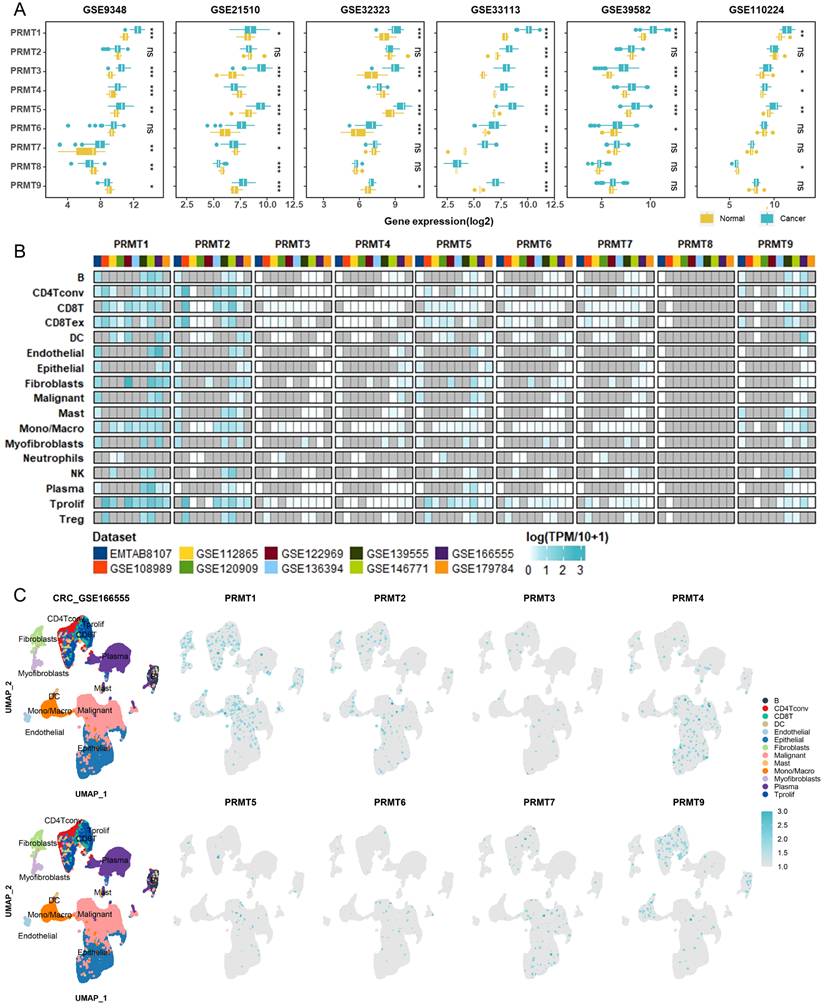
To validate our pan-cancer findings within a specific clinical context, we focused our investigation on CRC—a malignancy where PRMT-mediated epigenetic dysregulation is a recognized driver of progression. We first validated the expression profiles of PRMTs in CRC using multiple independent cohorts from the GEO database. Analysis of six GEO datasets revealed that PRMT1, 3, and 5 were consistently upregulated in tumor tissues (Figure 6A). In contrast, PRMT4 showed upregulation in four datasets but downregulation in two, while other PRMT members exhibited differential expression only in a subset of the cohorts (Figure 6A). Furthermore, to investigate the potential involvement of PRMTs in therapeutic response, we extended our analysis to six GEO datasets from patients receiving fluoropyrimidine-based chemotherapy, as well as three additional datasets involving bevacizumab treatment. Notably, significant differences in expression relative to treatment outcomes were observed only for PRMT3, 4, and 5 in a few specific datasets (Figures S10A-S10I), suggesting that PRMT expression levels may not be robustly associated with therapeutic efficacy. To address the cellular heterogeneity inherent in bulk transcriptomic data, we further investigated the single-cell expression patterns of PRMTs in CRC. Analysis of scRNA-seq data revealed that PRMT1 and PRMT2 were broadly distributed across immune, stromal, and malignant cell populations (Figures 6B, 6C). Collectively, these results indicate that although specific PRMTs (particularly PRMT1, 3, and 5) are consistently overexpressed in CRC, their transcript levels are not strong predictors of treatment response, and their widespread distribution suggests complex, multifaceted roles within the tumor microenvironment.
Validation of PRMT expression in CRC using bulk and single-cell transcriptomics. (A) Box plots showing the mRNA expression levels of PRMT family members across multiple independent CRC GEO cohorts. (B) Overview of PRMT expression across diverse cell populations in CRC scRNA-seq data. The color gradient represents the relative expression level, while gray indicates missing values or data not detected. (C) Scatter plots showing cell-type clustering and the expression of PRMT members in the GSE166555 dataset. Note that PRMT8 was excluded due to its absence in this dataset.
PRMT1 Is Upregulated in CRC and Promotes CRC Cell Growth in Real-World Validation
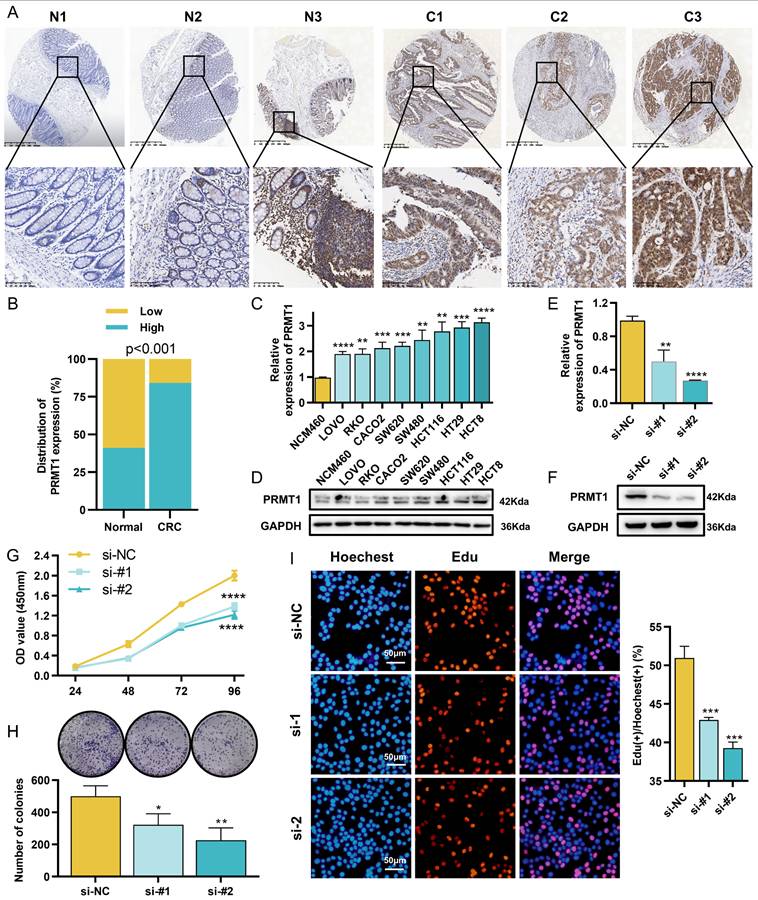
Given its elevated expression and functional dependency in CRC cells (Figures 1B, 6A-6C, S1A, S1B, S9), PRMT1 was prioritized for validation in a real-world clinical cohort. We first characterized the expression and spatial distribution of PRMT1 in paraffin-embedded tissues using IHC. In the normal mucosa, PRMT1 exhibited variable staining intensity across epithelial, stromal, and immune cells (Figure 7A). In contrast, PRMT1 expression was markedly higher in tumor cells compared to the relatively weak staining observed in the tumor stroma and infiltrating immune cells (Figure 7A). Regarding its subcellular localization, PRMT1 was predominantly restricted to the nucleus in both normal and malignant tissues (Figure 7A). Quantitative analysis revealed that the PRMT1 positivity rate was significantly higher in tumor tissues than in normal controls (Figure 7B). Furthermore, clinicopathological analysis demonstrated that high PRMT1 expression was significantly associated with larger tumor size, advanced T stage, and lymph node metastasis (Table 1). Consistent with these clinical findings, PRMT1 was found to be upregulated in CRC cell lines at both the mRNA and protein levels (Figures 7C, 7D).
PRMT1 is overexpressed in CRC and required for tumor cell growth. (A) Representative IHC staining of PRMT1 in CRC tissues (C1-C3) and adjacent normal mucosa (N1-N3). (B) Quantification of PRMT1 expression in the clinical cohort, shown as positivity rates. (C, D) Basal expression levels of PRMT1 in CRC cell lines detected by qRT-PCR (C) and Western blotting (D). (E, F) Validation of PRMT1 knockdown efficiency in HCT8 cells at the mRNA level (E) and protein level (F). (G-I) Effects of PRMT1 knockdown on HCT8 cell proliferation, assessed by CCK-8 assay (G), colony formation assay (H), and EdU proliferation assay (I).
Correlations between PRMT1 expression and clinicopathologic features in 120 CRC patients.
| Clinicopathological feature | Expression of PRMT1 | P value | ||
|---|---|---|---|---|
| Total (n=120) | Low (n=19) | High (n=101) | ||
| Gender | ||||
| Male | 83 | 12 | 71 | 0.584 |
| Femal | 37 | 7 | 30 | |
| Age | ||||
| < 60 | 67 | 10 | 57 | 0.803 |
| ≥ 60 | 53 | 9 | 44 | |
| Tumor size | ||||
| ≤ 5 cm | 79 | 17 | 62 | 0.021 |
| > 5 cm | 41 | 2 | 39 | |
| Tumor location | ||||
| Colon | 70 | 9 | 61 | 0.316 |
| Rectum | 50 | 10 | 40 | |
| Differentiation | ||||
| Well and Moderate | 107 | 18 | 89 | 1 |
| Poor | 13 | 1 | 12 | |
| T stage | ||||
| T1-2 | 15 | 11 | 4 | <0.001 |
| T3-4 | 105 | 8 | 97 | |
| Lymph node metastasis | ||||
| No | 73 | 16 | 57 | 0.027 |
| Yes | 47 | 3 | 44 | |
| Distant metastasis | ||||
| No | 115 | 19 | 96 | 1 |
| Yes | 5 | 0 | 5 | |
Aligning with our pan-cancer observations linking PRMT1 to cell growth, we performed further validation using GEO datasets. GSVA and GSEA identified a positive correlation between PRMT1 expression and pathways related to the cell cycle and DDR (Figures S11A, S11B). To experimentally investigate the functional role of PRMT1, we performed siRNA-mediated knockdown in HCT8 cells, achieving substantial depletion of PRMT1 (Figures 7E, 7F). Subsequent functional assays—including CCK-8 (Figure 7G), colony formation (Figure 7H), and EdU proliferation assay (Figure 7I)—demonstrated that PRMT1 silencing significantly impaired HCT8 cell proliferation. Collectively, these findings establish PRMT1 as a clinically relevant oncogene in CRC that correlates with disease progression and is essential for maintaining the proliferative capacity of tumor cells.
Discussion
PRMTs play crucial roles in cancer biology, regulating processes like gene expression, signal transduction, and DNA repair [9, 42, 43]. In this study, we systematically characterized the pan-cancer expression landscape of PRMTs using TCGA and GTEx datasets. Our findings reveal widespread dysregulation of PRMTs across various malignancies. Specifically, PRMT1, 3, 4, 5, 6, and 7 were predominantly upregulated, while PRMT2 and PRMT9 were consistently downregulated, suggesting divergent roles for these proteins in tumorigenesis. Mechanistically, we identified CNV-driven genomic instability as the primary driver of PRMT dysregulation, showing a strong positive correlation with mRNA levels. In contrast, DNA methylation exhibited a limited regulatory impact. Furthermore, the prognostic significance of PRMTs appeared highly context-dependent: high expression of PRMT1, 3, and 4 consistently predicted poor survival, supporting their roles as oncogenic drivers, whereas PRMT2, 5, 6, 7, and 9 showed tumor-specific prognostic effects. These findings suggest that the coordinated but distinct dysregulation of PRMT family members contributes to tumor progression in a context-specific manner.
The maintenance of genomic integrity is orchestrated by a sophisticated network of surveillance systems, including DNA damage checkpoints and repair machineries [44]. PRMT family members emerge as pivotal regulators in this context, directly impacting genomic stability through the modulation of DNA repair pathways [5]. Specifically, PRMT1, 4, and 5 facilitate homologous recombination by arginine methylating key repair factors, including MRE11, hnRNPUL1, and BRCA1 [45-47]. Moreover, PRMT1 and PRMT5 have been shown to influence non-homologous end joining through the regulation of 53BP1 [45, 48], while PRMT1, 5, and 6 contribute to base excision repair, highlighting their multifaceted contributions to genomic preservation [49, 50]. Consistent with these roles, our study reveals a strong correlation between the expression of PRMT1, 3, 4, and 5 and clinical markers of genomic damage (HRD, LOH, ploidy, and MSI). Enrichment analyses further underscore the involvement of PRMTs in cell proliferation and DDR pathways. Collectively, these results suggest that PRMT dysregulation fuels the accumulation of genomic heterogeneity by disrupting DNA repair, thereby propelling malignant transformation.
CSCs are fundamental orchestrators of tumor progression and treatment failure, owing to their robust self-renewal and plasticity [51]. Evidence indicates that PRMTs orchestrate CSC biological behavior through the arginine methylation of key transcription factors (e.g., OCT4, SOX2, and MYC) and the modulation of canonical oncogenic pathways, such as WNT and RAS signaling [5]. In the present study, we observed that PRMT expression levels significantly correlated with the transcriptomic stemness index (RNAss), while showing relatively weak associations with epigenetic stemness indices (e.g., DNAss and DMPss). This disparity suggests that PRMTs may modulate tumor plasticity primarily through post-transcriptional mechanisms, potentially by methylating key transcription factors or splicing regulators. Moreover, functional enrichment analysis validated the pervasive positive associations between PRMTs and key stemness-regulatory networks (e.g., Wnt, Hedgehog, and TGF-β). Taken together, these data highlight PRMTs as central regulators of tumor heterogeneity and malignant progression, providing a novel framework for precision-targeted interventions against the CSC niche.
The TME, an intricate ecosystem of immune cells, stroma, and extracellular matrix, maintains a reciprocal relationship with the cellular epigenome [52, 53]. While TME-derived physicochemical cues trigger epigenetic reprogramming, the subsequent epigenetic shifts in tumor and stromal cells feed back to reshape the TME, fueling malignancy and immune evasion [53]. As pivotal epigenetic modifiers, PRMTs are deeply involved in anti-tumor immune responses by modulating innate immune signaling pathways and the expression of key molecules such as PD-L1 and MHC-I [5]. Notably, targeting PRMT1 or PRMT5 has been shown to alleviate immunosuppression and sensitize tumors to PD-1/PD-L1 blockade [54, 55]. Here, we characterize the functional divergence of the PRMT family in TME remodeling. Upregulation of PRMT1, 3, and 5, alongside PRMT2 downregulation, characterizes “immune-excluded” or “cold” tumor profiles, indicating a synergistic contribution to immune surveillance escape. Conversely, PRMT4, 6, and 9 may indirectly modulate the TME through stromal expansion and angiogenesis, as evidenced by their positive correlation with fibroblast and endothelial infiltration despite negative associations with immune scores. The pronounced inter-cancer heterogeneity of PRMT-related immune landscapes underscores the need for precision oncology. While these findings highlight the PRMT-TME nexus, further in vivo and in vitro mechanistic studies are required. Overall, decoding the crosstalk between PRMTs and the TME provides a theoretical foundation for understanding immunoediting and optimizing combinatorial immunotherapy.
We selected CRC, a globally prevalent and highly heterogeneous malignancy, as a representative model to validate our pan-cancer findings [20, 56]. While PRMT1 and PRMT5 are known oncogenic drivers in CRC [14, 57], the collective impact of the PRMT family on CRC progression remains to be systematically elucidated. Our analysis of six independent GEO cohorts confirmed the consistent upregulation of PRMT1, 3, and 5, mirroring our pan-cancer results. Notably, in our clinical cohort, PRMT1 overexpression was positively associated with tumor size, T-stage, and nodal metastasis, highlighting its clinical relevance. This was further supported by functional evidence: both CRISPR-Cas9 screen data and in vitro silencing of PRMT1 demonstrated a significant suppression of CRC cell growth. Mechanistically, PRMT1 was found to be tightly linked to cell proliferation and DDR signaling, implying that PRMT1 supports CRC malignancy by maintaining genomic integrity and cell cycle progression. Collectively, these multi-dimensional insights—bridging bioinformatics, clinical data, and functional validation—identify PRMT1 as a central regulator of CRC growth and a promising target for precision therapy.
Despite the significant insights offered by this pan-cancer analysis of the PRMT family, certain limitations remain. A primary constraint is the reliance on public repositories (TCGA, GTEx, and GEO), where inter-cohort heterogeneity in sequencing methodologies and sample sizes may impact the robustness of our conclusions. To enhance the universality of these findings, future integration of multi-center clinical data is required. Furthermore, the scope of our functional validation was largely restricted to PRMT1 in the context of colorectal cancer. While these findings are promising, the mechanistic underpinnings of PRMT1 in other oncogenic contexts, as well as the roles of other PRMT family members, remain to be systematically elucidated. Prospective studies should encompass the full spectrum of the PRMT family across diverse cancer models. Lastly, the observed associations between PRMTs and the immune microenvironment necessitate further mechanistic confirmation. Investigating the translational potential of PRMTs as targets for combinatorial immunotherapy will be essential for advancing precision oncology and optimizing therapeutic strategies.
Conclusion
In summary, our multi-omic analysis demonstrates that the PRMT family is extensively dysregulated across cancers, driving genomic instability, cancer stemness, and immune evasion. Specifically, our validation of PRMT1 in CRC underscores its clinical significance and its critical role in tumor progression. By integrating pan-cancer data with experimental validation, this study establishes the PRMT family as a key therapeutic target and provides a foundation for personalized cancer treatment strategies.
Abbreviations
ACC: adrenocortical carcinoma; aDMA: asymmetric dimethylarginine; BLCA: bladder urothelial carcinoma; BRCA: breast invasive carcinoma; CHOL: cholangiocarcinoma; CNV: copy number variation; COAD: colon adenocarcinoma; CRC: colorectal cancer; DDR: DNA damage repair; DFI: disease-free interval; DLBCL: diffuse large B-cell lymphoma; DSS: disease-specific survival; ESCA: esophageal carcinoma; GBM: glioblastoma multiforme; GEO: gene expression omnibus; GSEA: gene set enrichment analysis; GTEx: genotype-tissue expression; HNSC: head and neck squamous cell carcinoma; HR: hazard ratio; HRD: homologous recombination deficiency; IF: immunofluorescence; IHC: immunohistochemistry; KICH: kidney chromophobe; KIRC: kidney renal clear cell carcinoma; LAML: acute myeloid leukemia; LGG: lower grade glioma; LIHC: liver hepatocellular carcinoma; LOH: loss of heterozygosity; LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; MATH: mutant-allele tumor heterogeneity; MMA: monomethylarginine; MSI: microsatellite instability; MSigDB: molecular signatures database; NES: normalized enrichment score; OS: overall survival; OV: ovarian serous cystadenocarcinoma; PFI: progression-free interval; PRMT: protein arginine methyltransferase; qRT-PCR: quantitative real-time polymerase chain reaction; SARC: sarcoma; scRNA-seq: single-cell RNA sequencing; sDMA: symmetric dimethylarginine; siRNAs: small interfering RNAs; SNVs: single nucleotide variants; STAD: stomach adenocarcinoma; TCGA: the cancer genome atlas; TGCT: testicular germ cell tumors; THCA: thyroid carcinoma; THYM: thymoma; TMA: tissue microarray; TMB: tumor mutational burden; TME: tumor microenvironment; UCEC: uterine corpus endometrial carcinoma; UCSC: the University of California, Santa Cruz.
Supplementary Material
Supplementary figures.
Acknowledgements
We sincerely thank Dr. Chunlin Ou from Xiangya Hospital for his assistance in the preparation of the TMA. We are grateful to the contributors of the GEO, GTEx, and TCGA databases for providing the invaluable datasets that made this study possible. Additionally, we acknowledge the use of Large Language Models (ChatGPT and Gemini) for linguistic refinement and proofreading during the preparation of this manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (Grant No.82403198), the Science and Technology Commission of Shanghai Municipality (24ZR1447300), and Natural Science Foundation of Hunan Province of China (Project No. 2025JJ60738).
Availability of data and materials
The datasets supporting the findings of this study are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
This study was reviewed and approved by the Ethics Committee of Xiangya Hospital, Central South University, in full accordance with the principles of the Declaration of Helsinki.
Author contributions
The conceptualization and data analysis were undertaken collaboratively by JX, QW, YX, XF and YL. YX, QW, and JX were responsible for drafting the manuscript, while YL and QW provided financial support for the study. Manuscript editing and revisions were carried out by JX, YX, YL, JL, XW, and YW. All authors have critically reviewed and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374-403
2. Graham TA, Sottoriva A. Measuring cancer evolution from the genome. J Pathol. 2017;241:183-91
3. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13:877-919
4. Bates SE. Epigenetic Therapies for Cancer. N Engl J Med. 2020;383:650-63
5. Xu Y, Wu Q, Zhang Y, Gu Y, Zhu H, Fu X. et al. Arginine methylation in cancer: mechanisms and therapeutic implications. Biomark Res. 2025;13:143
6. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37-50
7. Fan Y, Wang Y, Dan W, Zhang Y, Nie L, Ma Z. et al. PRMT5-mediated arginine methylation stabilizes GPX4 to suppress ferroptosis in cancer. Nat Cell Biol. 2025;27:641-53
8. Meng L, Jiang Y, You J, Chen Y, Guo S, Chen L. et al. PRMT1-methylated MSX1 phase separates to control palate development. Nat Commun. 2025;16:949
9. Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol. 2019;20:642-57
10. Fulton MD, Brown T, Zheng YG. Mechanisms and Inhibitors of Histone Arginine Methylation. Chem Rec. 2018;18:1792-807
11. Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1-13
12. Gao Y, Feng C, Ma J, Yan Q. Protein arginine methyltransferases (PRMTs): Orchestrators of cancer pathogenesis, immunotherapy dynamics, and drug resistance. Biochem Pharmacol. 2024;221:116048
13. Hsu WJ, Chiang MC, Chao YC, Chang YC, Hsu MC, Chung CH. et al. Arginine Methylation of DDX3 by PRMT1 Mediates Mitochondrial Homeostasis to Promote Breast Cancer Metastasis. Cancer Res. 2024;84:3023-43
14. Yao B, Gui T, Zeng X, Deng Y, Wang Z, Wang Y. et al. PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med. 2021;13:58
15. Peng L, Zhao Y, Tan J, Hou J, Jin X, Liu DX. et al. PRMT1 promotes Warburg effect by regulating the PKM2/PKM1 ratio in non-small cell lung cancer. Cell Death Dis. 2024;15:504
16. Walton J, Ng ASN, Arevalo K, Apostoli A, Meens J, Karamboulas C. et al. PRMT1 inhibition perturbs RNA metabolism and induces DNA damage in clear cell renal cell carcinoma. Nat Commun. 2024;15:8232
17. Li Y, Dou Y, Da Veiga Leprevost F, Geffen Y, Calinawan AP, Aguet F. et al. Proteogenomic data and resources for pan-cancer analysis. Cancer Cell. 2023;41:1397-406
18. Wang J, Wu M, Sun J, Chen M, Zhang Z, Yu J. et al. Pan-cancer analysis identifies protein arginine methyltransferases PRMT1 and PRMT5 and their related signatures as markers associated with prognosis, immune profile, and therapeutic response in lung adenocarcinoma. Heliyon. 2023;9:e22088
19. Qiu Y, Wang H, Liao P, Xu B, Hu R, Yang Y. et al. Systematic pan-cancer landscape identifies CARM1 as a potential prognostic and immunological biomarker. BMC Genom Data. 2022;23:7
20. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17-48
21. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
22. Benson AB, Venook AP, Al-Hawary MM, Arain MA, Chen YJ, Ciombor KK. et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021;19:329-59
23. Benson AB, Venook AP, Al-Hawary MM, Azad N, Chen YJ, Ciombor KK. et al. Rectal Cancer, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2022;20:1139-67
24. Saez MA, Garcia-Montero C, Fraile-Martinez O, Minaya-Bravo AM, Boaru DL, De Leon-Oliva D. et al. Differential expression of ferroptosis markers, circadian regulators, KLOTHO, and classical tumor suppressors in colorectal cancer according to tumor stage: Influence of age, anatomical location, and correlation patterns. Histol Histopathol. 2025;40:1985-2009
25. Cendra AS, Pekarek L, Ospino LR, Dbouk Y, Chnaiker S, Luengo A. et al. Predictive and Prognostic Biomarkers of Recurrence in Locoregional Colorectal Cancer. J Cancer. 2025;16:3024-39
26. Cao M, Shao Z, Qian X, Chen M, Deng C, Chen X. et al. TRIM21-mediated PRMT1 degradation attenuates colorectal cancer malignant progression. Cell Death Dis. 2025;16:56
27. Cheng C, Zhang K, Lu M, Zhang Y, Wang T, Zhang Y. RPF2 and CARM1 cooperate to enhance colorectal cancer metastasis via the AKT/GSK-3beta signaling pathway. Exp Cell Res. 2025;444:114374
28. Zhang X, Jin M, Chu Y, Liu F, Qu H, Chen C. PRMT6 promotes colorectal cancer progress via activating MYC signaling. J Transl Med. 2025;23:74
29. Zou H, Liu Y, Yang X, Zhang Q, Pan Q, Huang J. et al. PRMT2 promotes tumorigenic phenotypes through the Wnt signaling pathway and drives immune suppression in Colorectal cancer. Cancer Lett. 2025;632:217967
30. Qu S, Feng B, Xing M, Qiu Y, Ma L, Yang Z. et al. PRMT5 K240lac confers ferroptosis resistance via ALKBH5/SLC7A11 axis in colorectal cancer. Oncogene. 2025;44:2814-30
31. Plotnikov A, Kozer N, Cohen G, Carvalho S, Duberstein S, Almog O. et al. PRMT1 inhibition induces differentiation of colon cancer cells. Sci Rep. 2020;10:20030
32. Abumustafa W, Zamer BA, Khalil BA, Hamad M, Maghazachi AA, Muhammad JS. Protein arginine N-methyltransferase 5 in colorectal carcinoma: Insights into mechanisms of pathogenesis and therapeutic strategies. Biomed Pharmacother. 2022;145:112368
33. Zhu J, Fu S, Zou X, Zeng H, Cui G, Peng Y. et al. PRMT5 Inhibitor Synergizes with Chemotherapy to Induce Resembling Mismatch Repair Deficiency and Enhance Anti-TIGIT Therapy in Microsatellite-Stable Colorectal Cancer. Adv Sci (Weinh). 2025;12:e2500271
34. Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2021;2:100141
35. Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612
36. Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218
37. Zeng D, Ye Z, Shen R, Yu G, Wu J, Xiong Y. et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front Immunol. 2021;12:687975
38. Li Y, Gan Y, Liu J, Li J, Zhou Z, Tian R. et al. Downregulation of MEIS1 mediated by ELFN1-AS1/EZH2/DNMT3a axis promotes tumorigenesis and oxaliplatin resistance in colorectal cancer. Signal Transduct Target Ther. 2022;7:87
39. Fan T, Zhang M, Yang J, Zhu Z, Cao W, Dong C. Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal Transduct Target Ther. 2023;8:450
40. Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H. et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779-84
41. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS. et al. Defining a Cancer Dependency Map. Cell. 2017;170:564-76 e16
42. Jarrold J, Davies CC. PRMTs and Arginine Methylation: Cancer's Best-Kept Secret? Trends Mol Med. 2019;25:993-1009
43. Zhu Y, Xia T, Chen DQ, Xiong X, Shi L, Zuo Y. et al. Promising role of protein arginine methyltransferases in overcoming anti-cancer drug resistance. Drug Resist Updat. 2024;72:101016
44. Requesens M, Foijer F, Nijman HW, de Bruyn M. Genomic instability as a driver and suppressor of anti-tumor immunity. Front Immunol. 2024;15:1462496
45. Vadnais C, Chen R, Fraszczak J, Yu Z, Boulais J, Pinder J. et al. GFI1 facilitates efficient DNA repair by regulating PRMT1 dependent methylation of MRE11 and 53BP1. Nat Commun. 2018;9:1418
46. Gurunathan G, Yu Z, Coulombe Y, Masson JY, Richard S. Arginine methylation of hnRNPUL1 regulates interaction with NBS1 and recruitment to sites of DNA damage. Sci Rep. 2015;5:10475
47. Montenegro MF, Gonzalez-Guerrero R, Sanchez-Del-Campo L, Pinero-Madrona A, Cabezas-Herrera J, Rodriguez-Lopez JN. PRMT1-dependent methylation of BRCA1 contributes to the epigenetic defense of breast cancer cells against ionizing radiation. Sci Rep. 2020;10:13275
48. Clarke TL, Sanchez-Bailon MP, Chiang K, Reynolds JJ, Herrero-Ruiz J, Bandeiras TM. et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol Cell. 2017;65:900-16 e7
49. Zhang Y, Zhang Q, Li L, Mu D, Hua K, Ci S. et al. Arginine methylation of APE1 promotes its mitochondrial translocation to protect cells from oxidative damage. Free Radic Biol Med. 2020;158:60-73
50. El-Andaloussi N, Valovka T, Toueille M, Steinacher R, Focke F, Gehrig P. et al. Arginine methylation regulates DNA polymerase beta. Mol Cell. 2006;22:51-62
51. Zhou G, Latchoumanin O, Bagdesar M, Hebbard L, Duan W, Liddle C. et al. Aptamer-Based Therapeutic Approaches to Target Cancer Stem Cells. Theranostics. 2017;7:3948-61
52. Elhanani O, Ben-Uri R, Keren L. Spatial profiling technologies illuminate the tumor microenvironment. Cancer Cell. 2023;41:404-20
53. Xie Z, Zhou Z, Yang S, Zhang S, Shao B. Epigenetic regulation and therapeutic targets in the tumor microenvironment. Mol Biomed. 2023;4:17
54. He Q, Zhang Y, Li W, Chen S, Xiong J, Zhao R. et al. Inhibition of PRMT5 moderately suppresses prostate cancer growth in vivo but enhances its response to immunotherapy. Cancer Lett. 2024;602:217214
55. Tao H, Jin C, Zhou L, Deng Z, Li X, Dang W. et al. PRMT1 Inhibition Activates the Interferon Pathway to Potentiate Antitumor Immunity and Enhance Checkpoint Blockade Efficacy in Melanoma. Cancer Res. 2024;84:419-33
56. Li J, Ma X, Chakravarti D, Shalapour S, DePinho RA. Genetic and biological hallmarks of colorectal cancer. Genes Dev. 2021;35:787-820
57. Liao HW, Hsu JM, Xia W, Wang HL, Wang YN, Chang WC. et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest. 2015;125:4529-43
Author contact
![]() Corresponding authors: Yimin Li, Department of Pathology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, No. 197, Ruijin Er Road, Huangpu District, Shanghai, China; Email: yimin_li_0107com; lym12999com.cn. Xiaodan Fu, Department of Pathology, Xiangya Hospital, Central South University, No.87 Xiangya Road of Changsha, China; Email: jessicafu0225com; fuxiaodanedu.cn.
Corresponding authors: Yimin Li, Department of Pathology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, No. 197, Ruijin Er Road, Huangpu District, Shanghai, China; Email: yimin_li_0107com; lym12999com.cn. Xiaodan Fu, Department of Pathology, Xiangya Hospital, Central South University, No.87 Xiangya Road of Changsha, China; Email: jessicafu0225com; fuxiaodanedu.cn.